

Computational Estimation of Microsecond to Second Atomistic Folding Times
- PMID: 30892023
- PMCID: PMC6660137
- DOI: 10.1021/jacs.8b10735
Computational Estimation of Microsecond to Second Atomistic Folding Times
Erratum in
-
Correction to "Computational Estimation of Microsecond to Second Atomistic Folding Times".J Am Chem Soc. 2021 May 19;143(19):7588. doi: 10.1021/jacs.0c02854. Epub 2021 May 7. J Am Chem Soc. 2021. PMID: 33961428 No abstract available.
Abstract
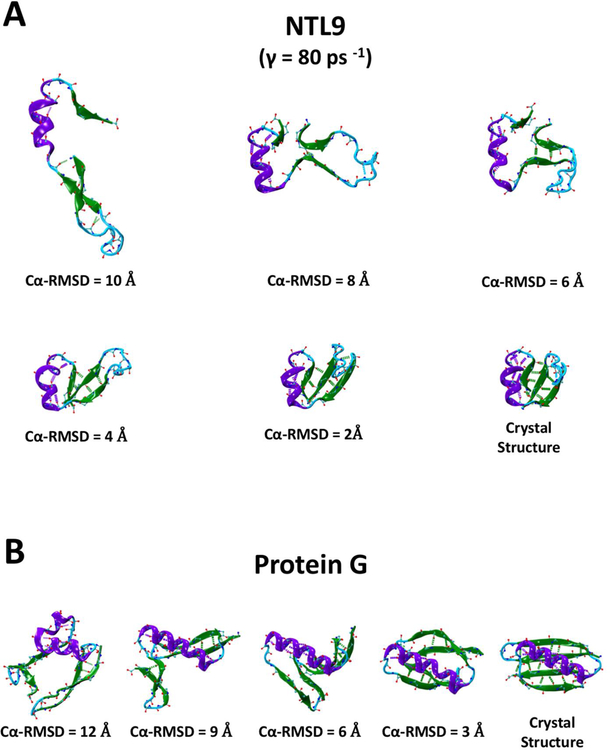
Despite the development of massively parallel computing hardware including inexpensive graphics processing units (GPUs), it has remained infeasible to simulate the folding of atomistic proteins at room temperature using conventional molecular dynamics (MD) beyond the microsecond scale. Here, we report the folding of atomistic, implicitly solvated protein systems with folding times τ ranging from ∼10 μs to ∼100 ms using the weighted ensemble (WE) strategy in combination with GPU computing. Starting from an initial structure or set of structures, WE organizes an ensemble of GPU-accelerated MD trajectory segments via intermittent pruning and replication events to generate statistically unbiased estimates of rate constants for rare events such as folding; no biasing forces are used. Although the variance among atomistic WE folding runs is significant, multiple independent runs are used to reduce and quantify statistical uncertainty. Folding times are estimated directly from WE probability flux and from history-augmented Markov analysis of the WE data. Three systems were examined: NTL9 at low solvent viscosity (yielding τf = 0.8-9 μs), NTL9 at water-like viscosity (τf = 0.2-2 ms), and Protein G at low viscosity (τf = 3-200 ms). In all cases, the folding time, uncertainty, and ensemble properties could be estimated from WE simulation; for Protein G, this characterization required significantly less overall computing than would be required to observe a single folding event with conventional MD simulations. Our results suggest that the use and calibration of force fields and solvent models for precise estimation of kinetic quantities is becoming feasible.
Figures
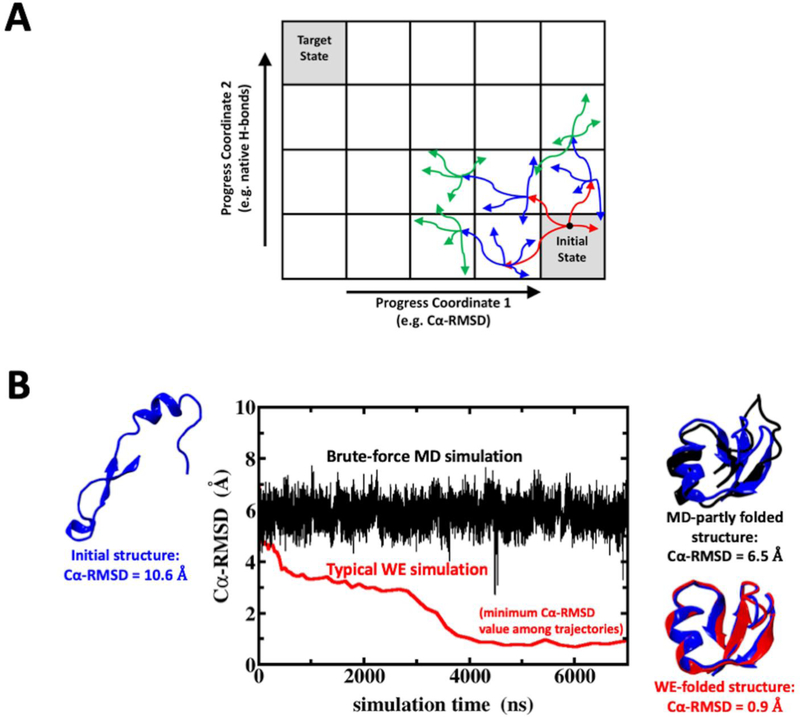
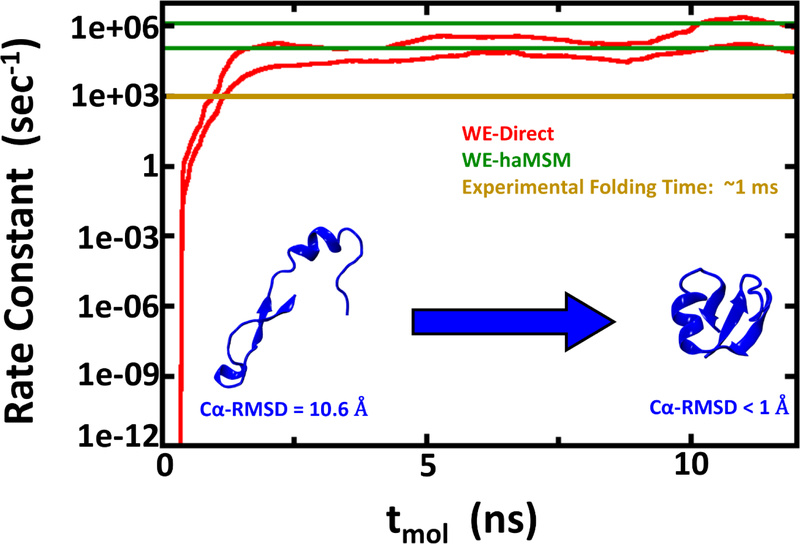
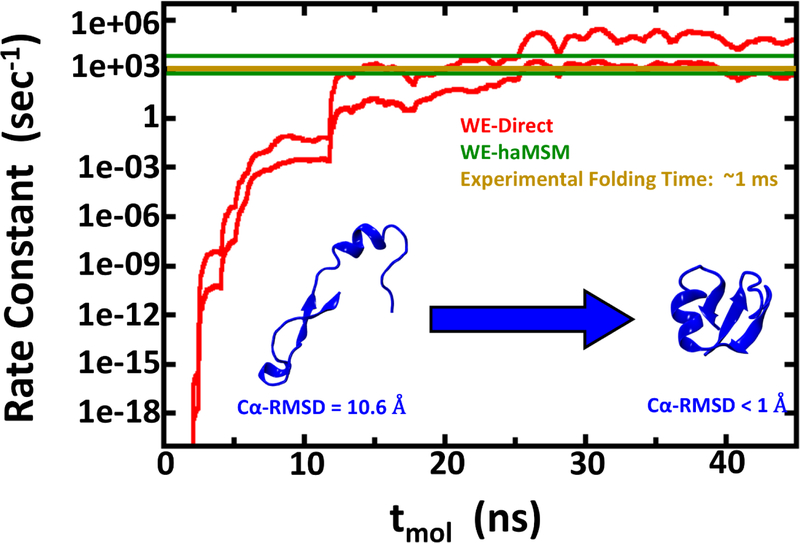
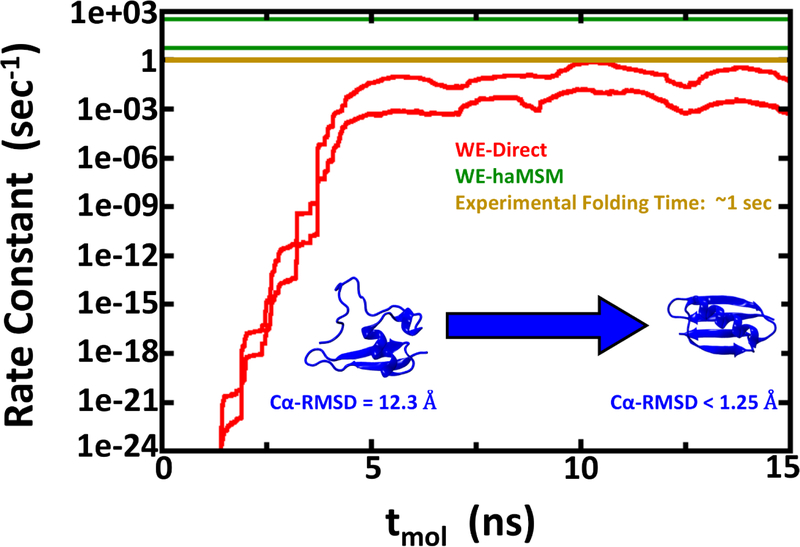
References
-
- Lindorff-Larsen K; Piana S; Dror RO; Shaw DE, How Fast-Folding Proteins Fold. Science 2011, 334 (6055), 517–520. - PubMed
-
- Wolynes PG; Eaton WA, The physics of protein folding. Physics World 1999, 12 (9), 39–44.
-
- Go N, Theoretical Studies of Protein Folding. Annual Review of Biophysics and Bioengineering 1983, 12 (1), 183–210. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
