

Reduced signal for polygenic adaptation of height in UK Biobank
- PMID: 30895923
- PMCID: PMC6428572
- DOI: 10.7554/eLife.39725
Reduced signal for polygenic adaptation of height in UK Biobank
Abstract
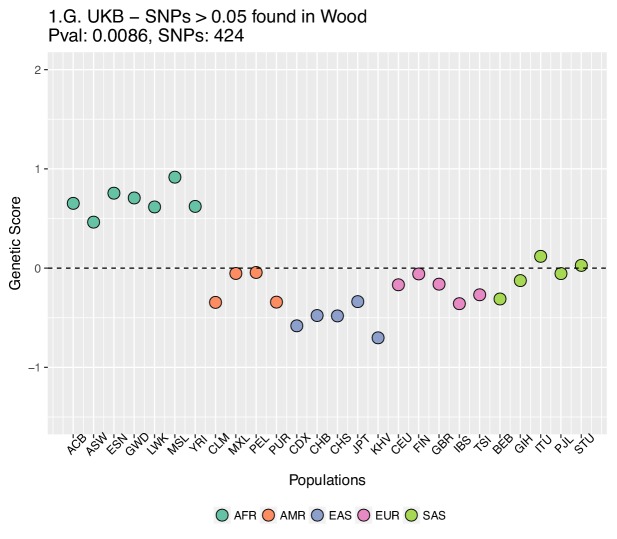
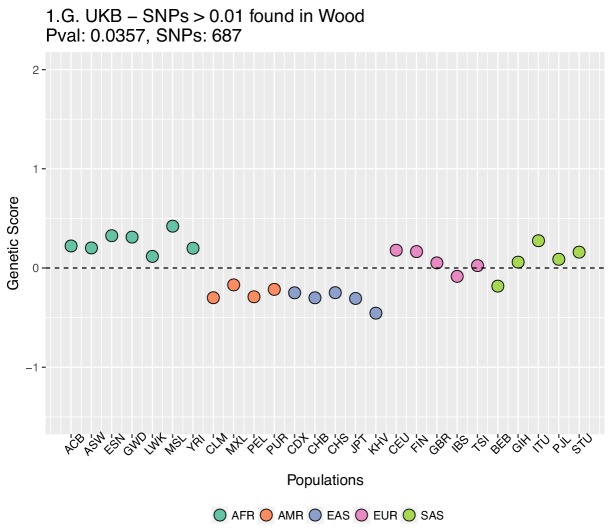
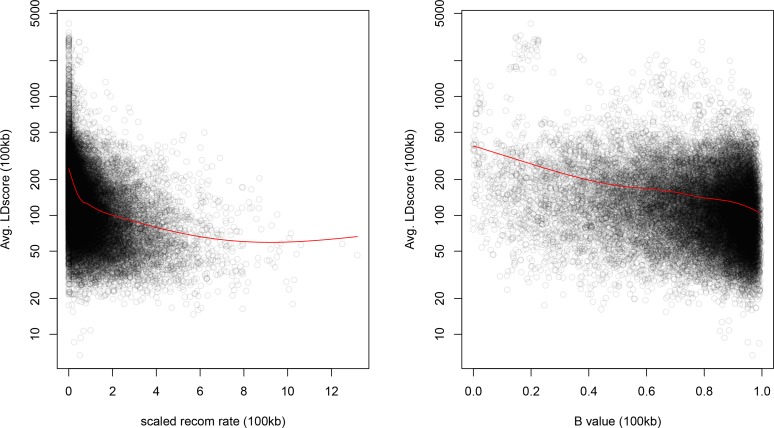
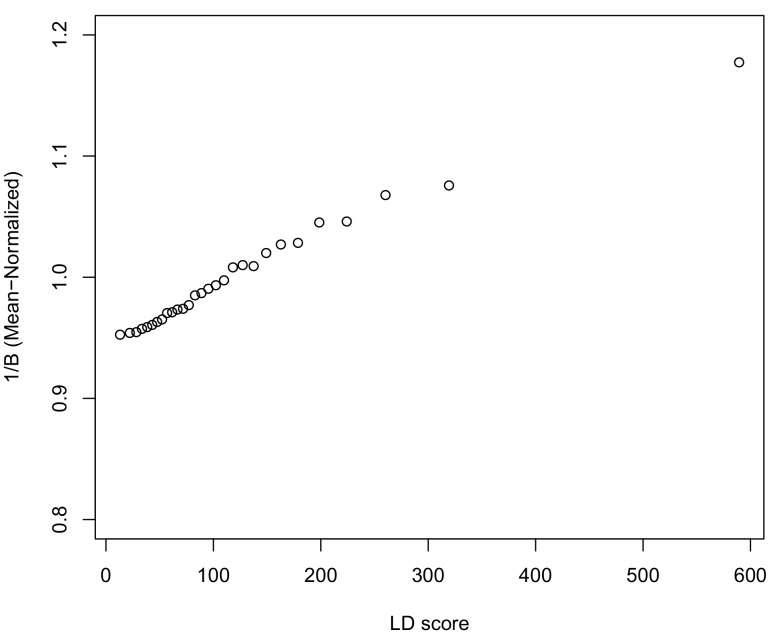
Several recent papers have reported strong signals of selection on European polygenic height scores. These analyses used height effect estimates from the GIANT consortium and replication studies. Here, we describe a new analysis based on the the UK Biobank (UKB), a large, independent dataset. We find that the signals of selection using UKB effect estimates are strongly attenuated or absent. We also provide evidence that previous analyses were confounded by population stratification. Therefore, the conclusion of strong polygenic adaptation now lacks support. Moreover, these discrepancies highlight (1) that methods for correcting for population stratification in GWAS may not always be sufficient for polygenic trait analyses, and (2) that claims of differences in polygenic scores between populations should be treated with caution until these issues are better understood.
Editorial note: This article has been through an editorial process in which the authors decide how to respond to the issues raised during peer review. The Reviewing Editor's assessment is that all the issues have been addressed (see decision letter).
Keywords: GWAS; evolutionary biology; genetics; genomics; human; natural selection; polygenic adaptation; population genetics; population structure; quantitative genetics.
© 2019, Berg et al.
Conflict of interest statement
JB, AH, NS, AJ, HM, YF, EB, XZ, FR, JP, GC No competing interests declared
Figures
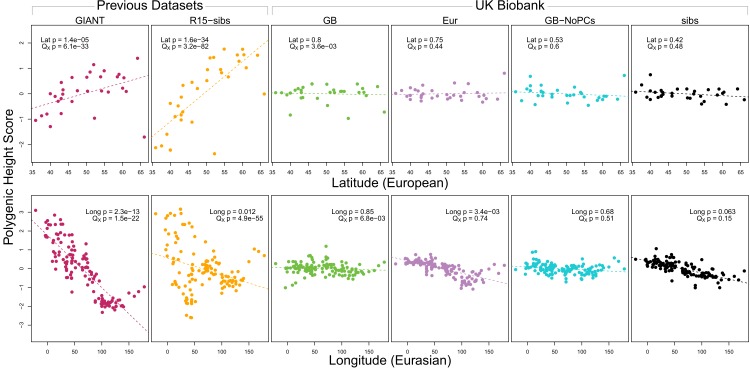
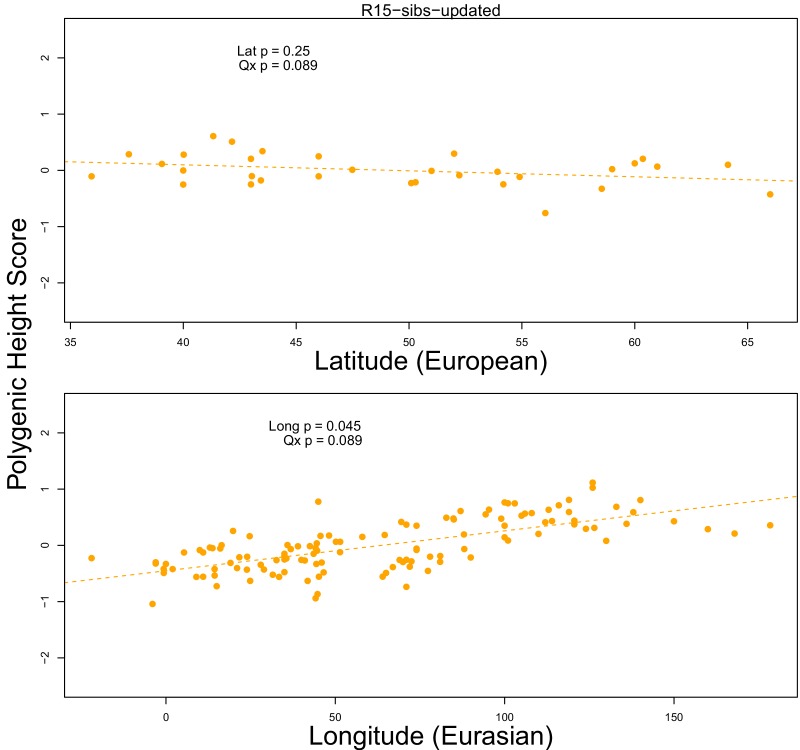
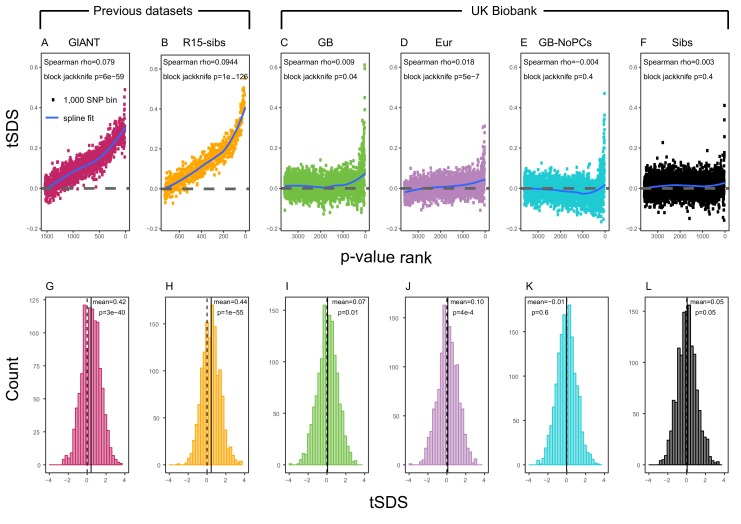
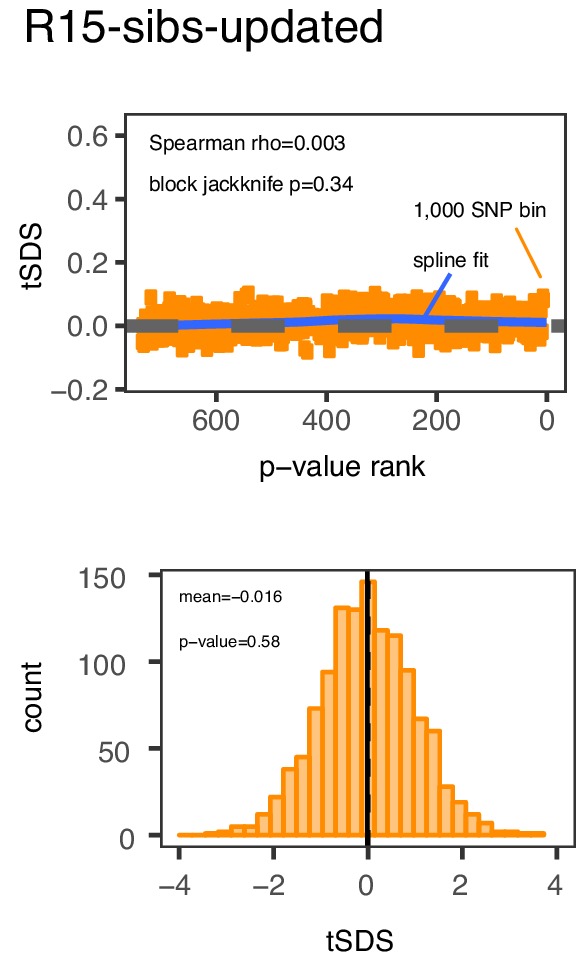
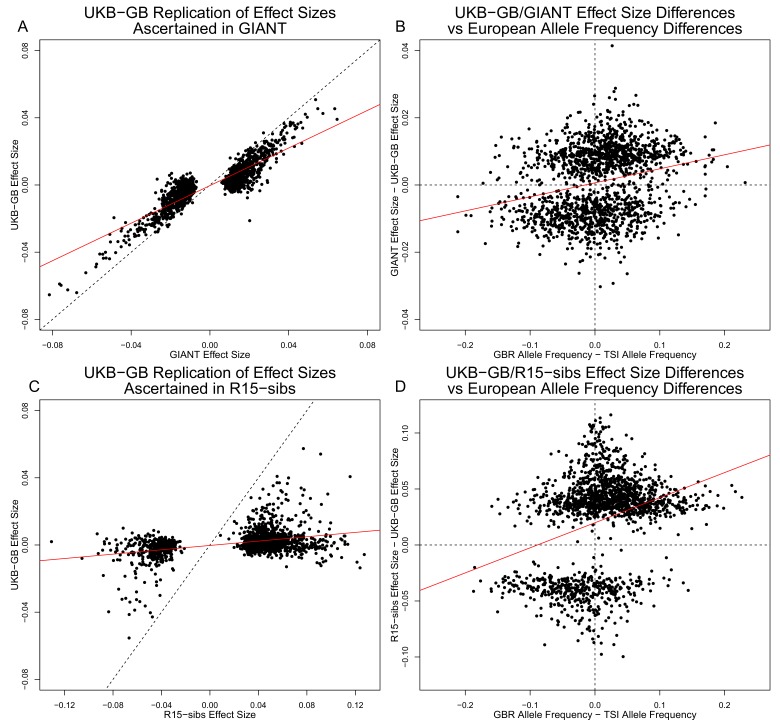
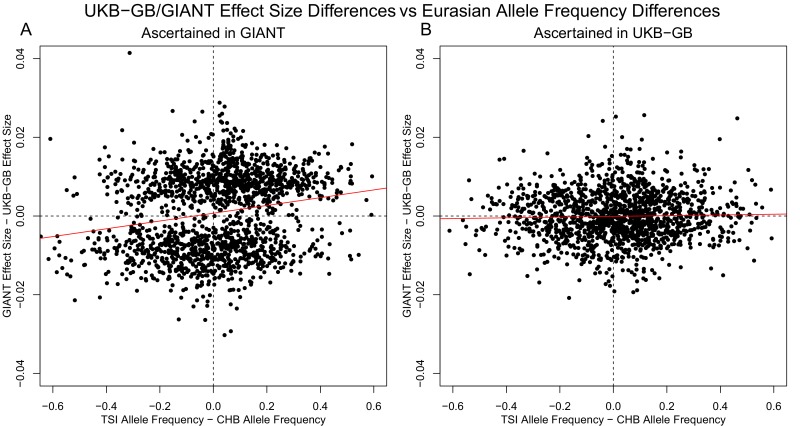
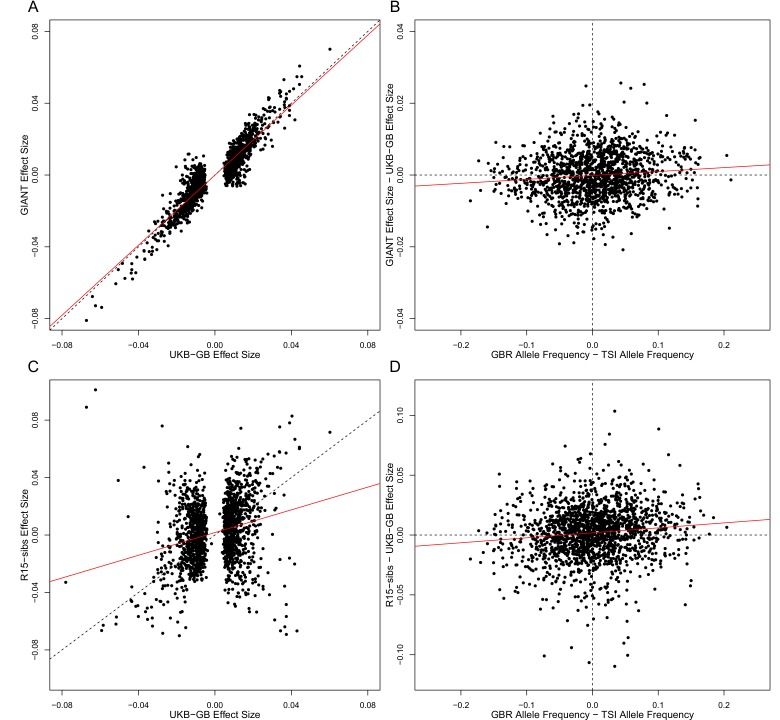
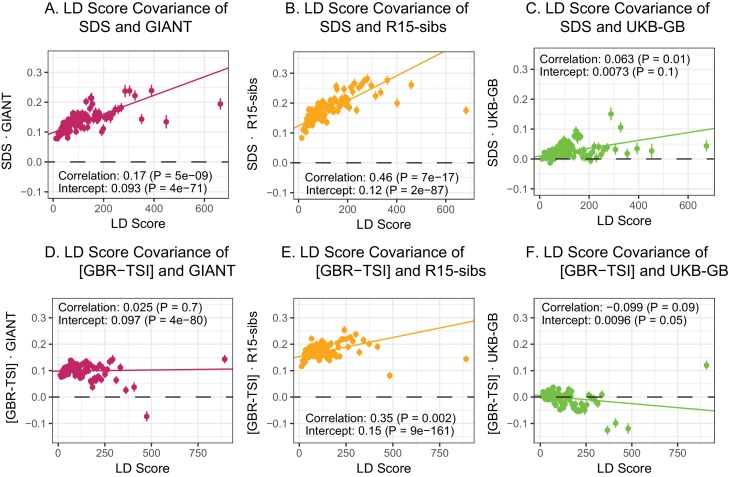
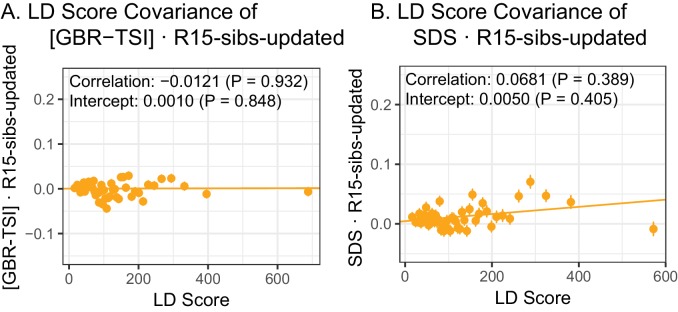
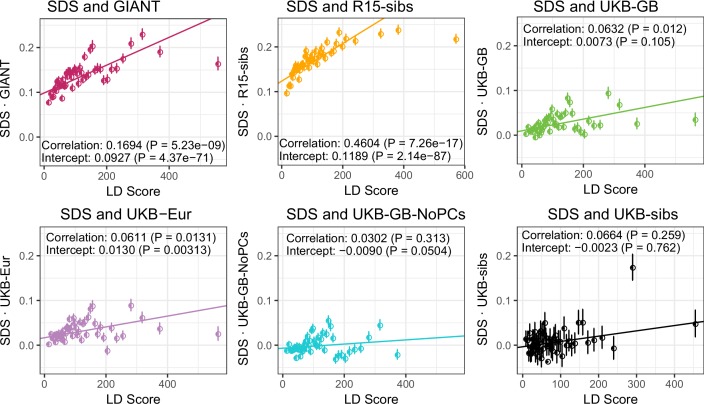
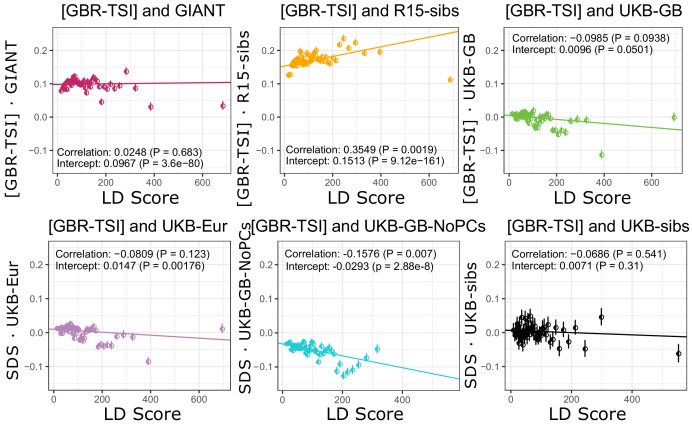
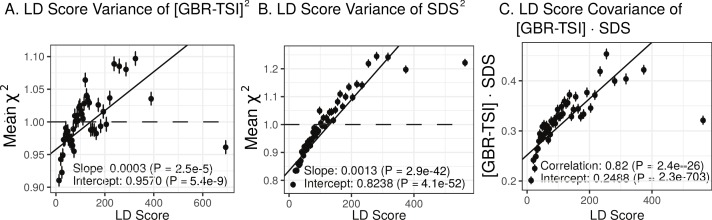
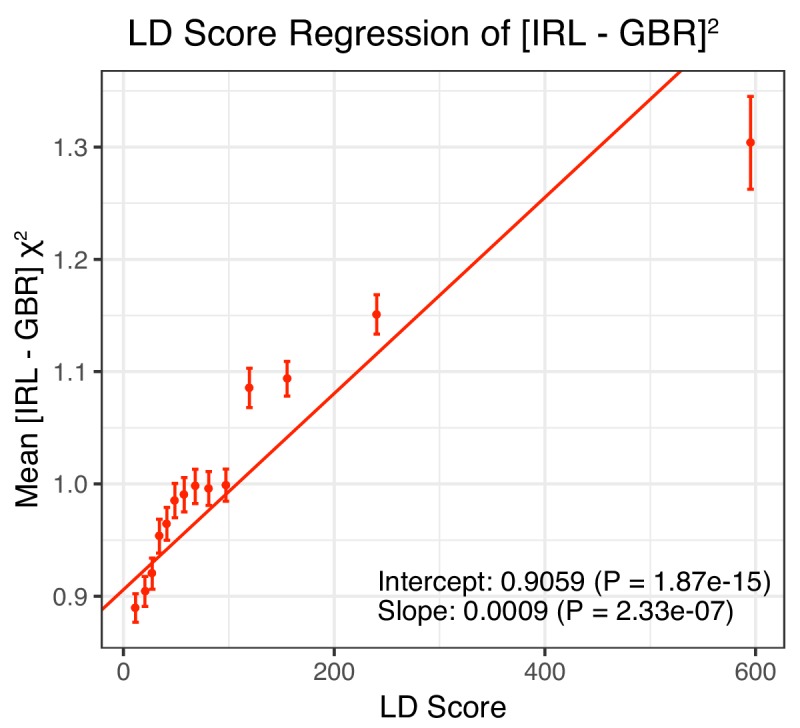
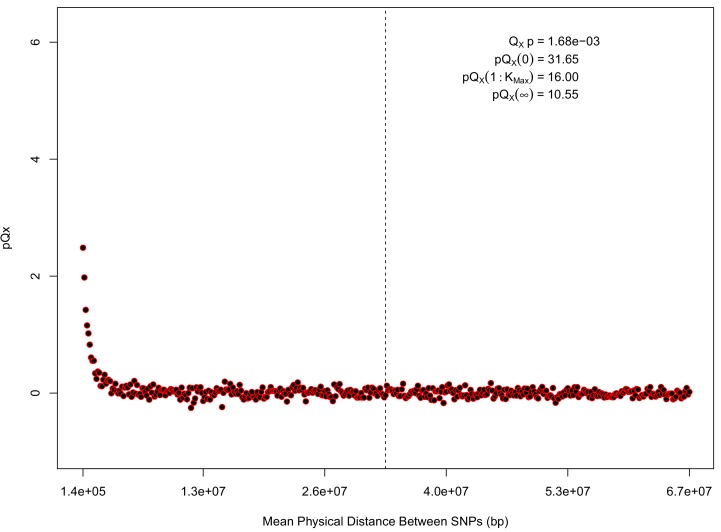
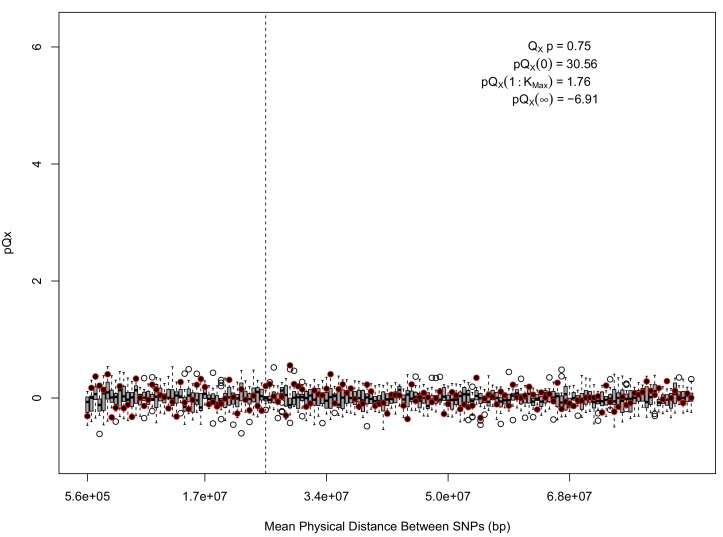
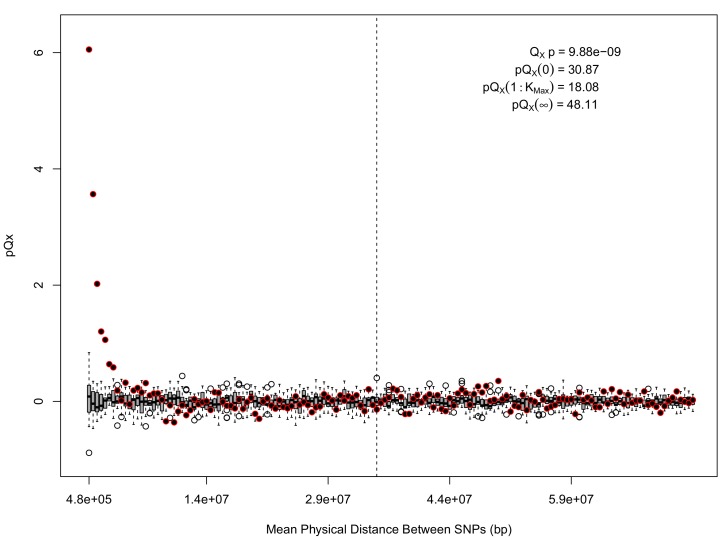
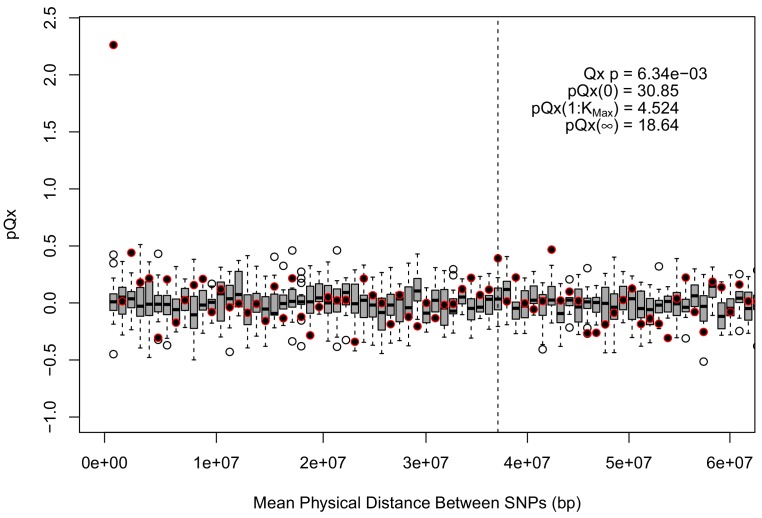
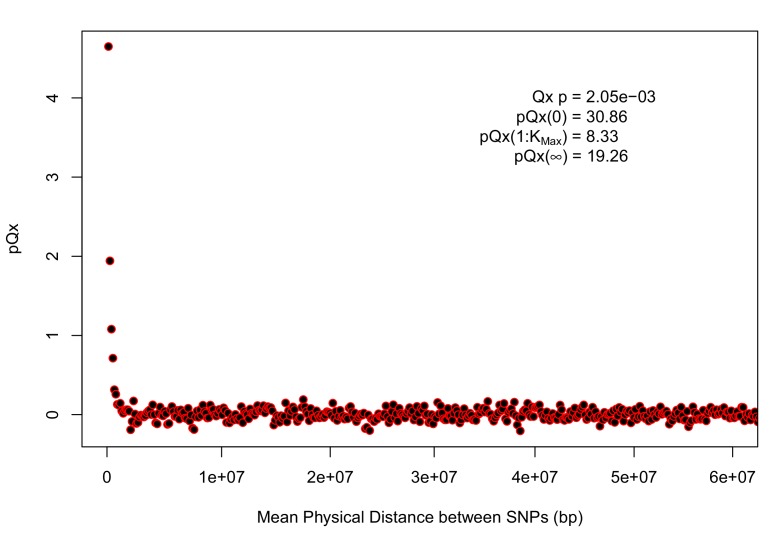
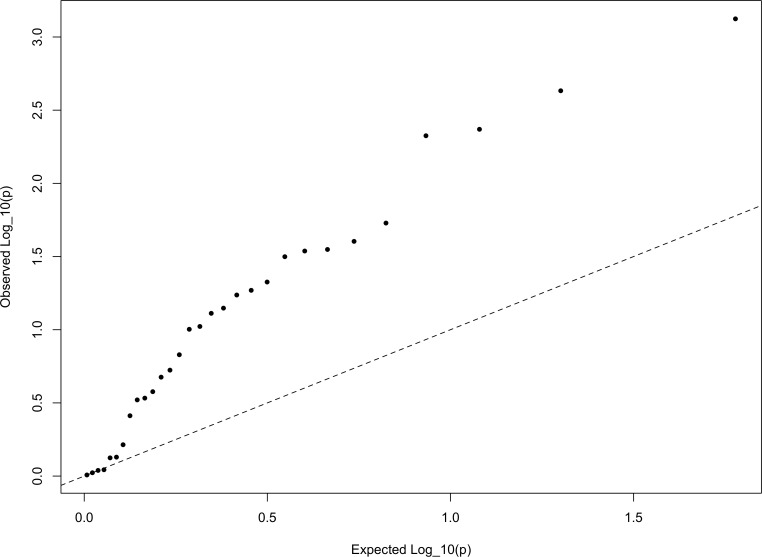
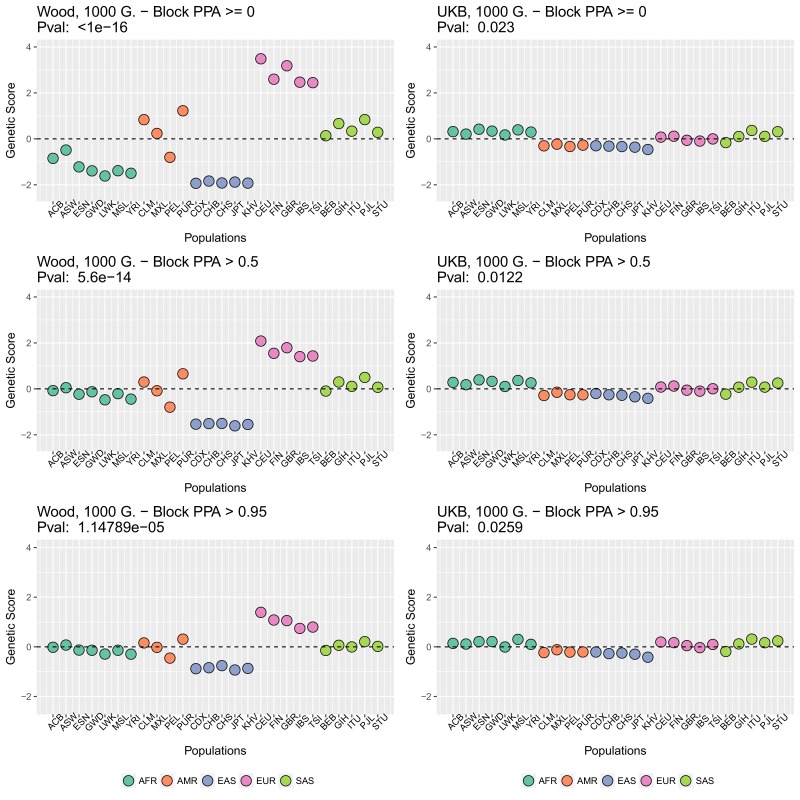
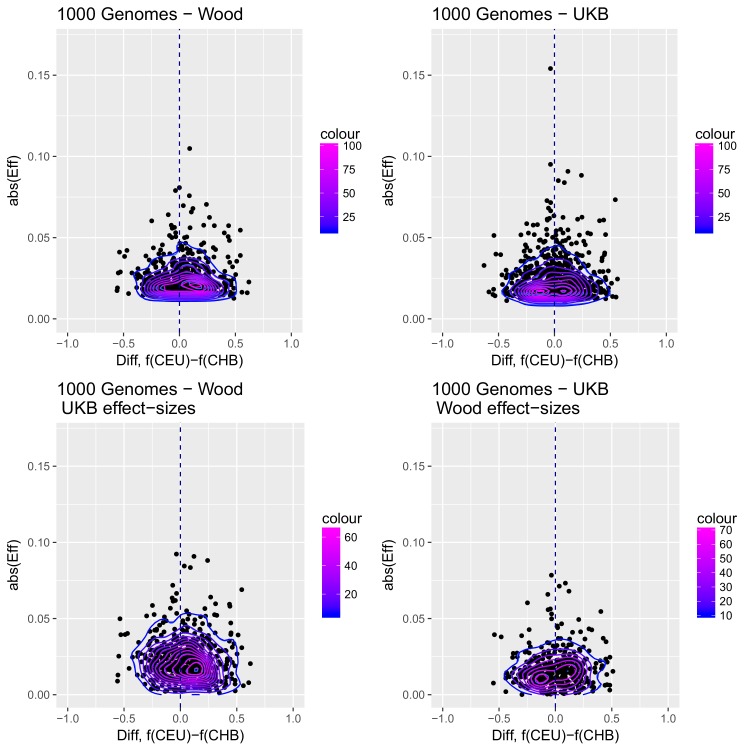
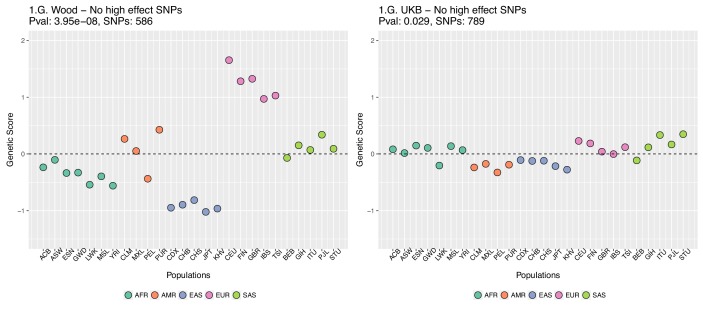
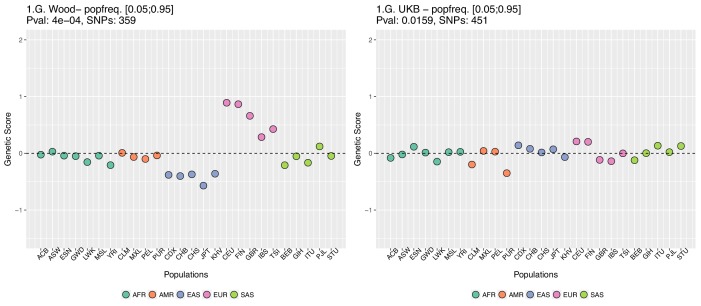
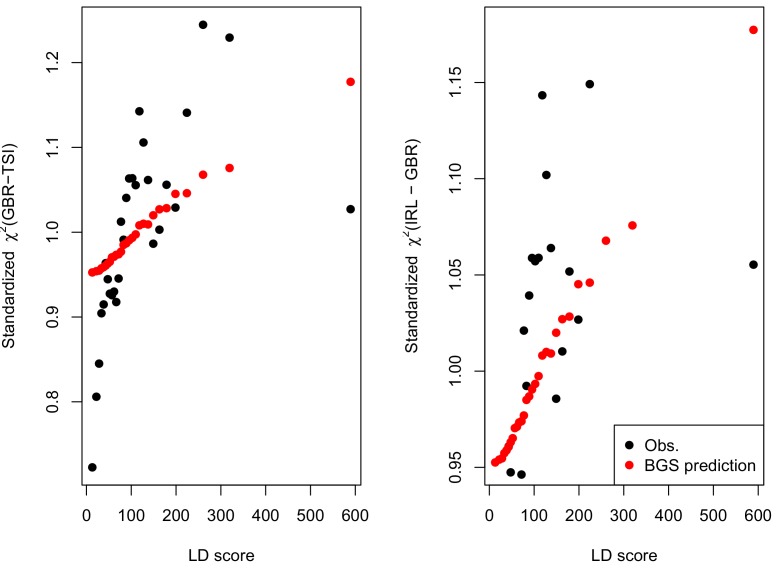
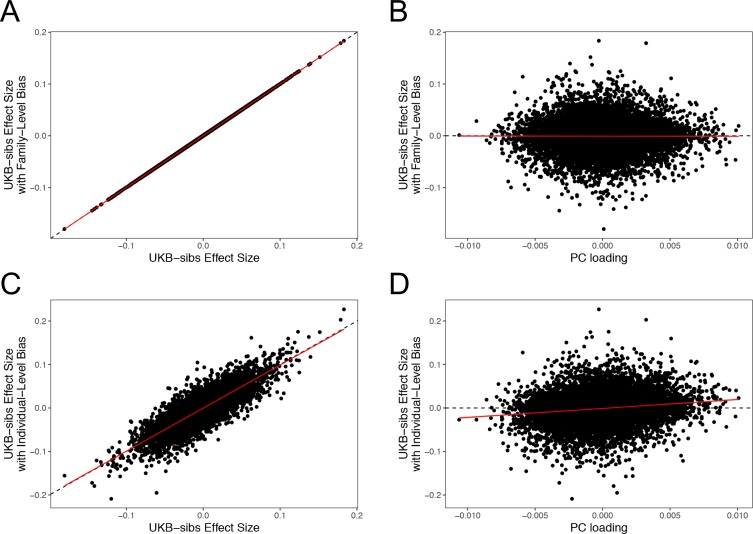
Comment in
-
Why structure matters.Elife. 2019 Mar 21;8:e45380. doi: 10.7554/eLife.45380. Elife. 2019. PMID: 30895925 Free PMC article.
References
-
- Berg JJ, Zhang X, Coop G. Polygenic adaptation has impacted multiple anthropometric traits. bioRxiv. 2017 doi: 10.1101/167551. - DOI
Publication types
MeSH terms
Associated data
Grants and funding
- Fellowship/Stanford Center for Computational, Evolutionary and Human Genomics/International
- R01 HG008140/HG/NHGRI NIH HHS/United States
- F32 GM126787/NH/NIH HHS/United States
- R01 GM115889/NH/NIH HHS/United States
- R01 GM121372/GM/NIGMS NIH HHS/United States
- Graduate Fellowship/Stanford University/International
- National Defense Science and Engineering Gran/U.S. Department of Defense/International
- F32 GM126787/GM/NIGMS NIH HHS/United States
- R01 GM108779/GM/NIGMS NIH HHS/United States
- National Defense Science and Engineering Grant/U.S. Department of Defense/International
- T32 HG000044/HG/NHGRI NIH HHS/United States
- R01 GM121372/NH/NIH HHS/United States
- R01 GM108779/NH/NIH HHS/United States
- Young Investigator award/Villum Fonden/International
- R01 HG008140/NH/NIH HHS/United States
- R01 GM115889/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
