

Mechanisms underlying aflatoxin-associated mutagenesis - Implications in carcinogenesis
- PMID: 30897375
- PMCID: PMC6959417
- DOI: 10.1016/j.dnarep.2019.03.004
Mechanisms underlying aflatoxin-associated mutagenesis - Implications in carcinogenesis
Abstract
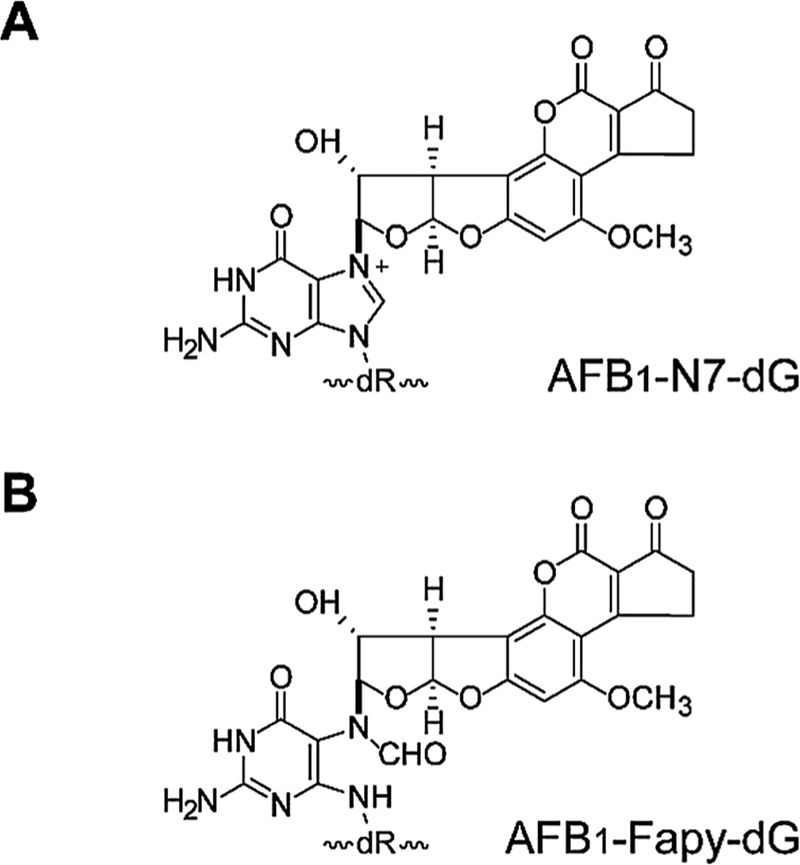
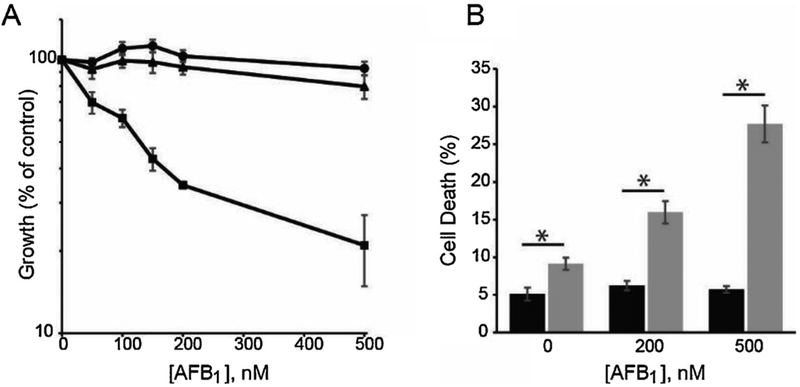
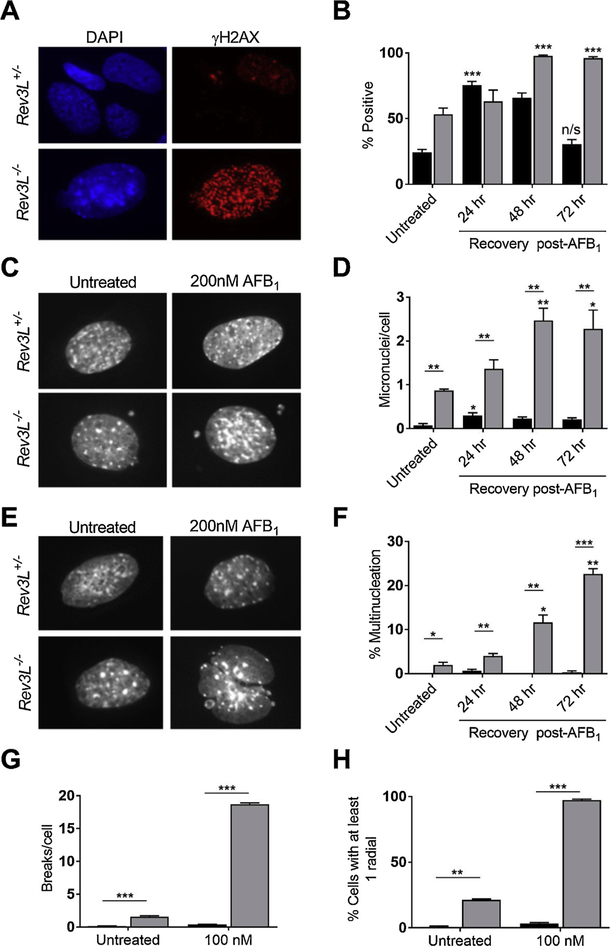
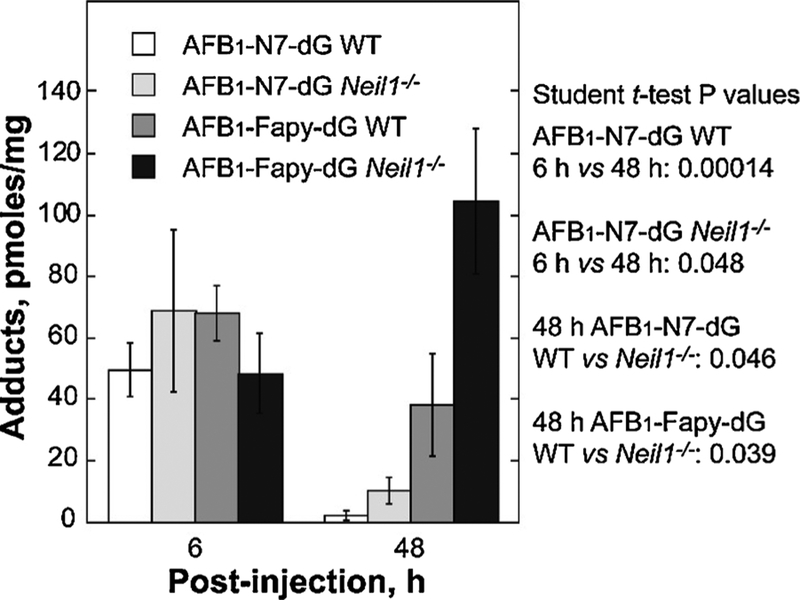
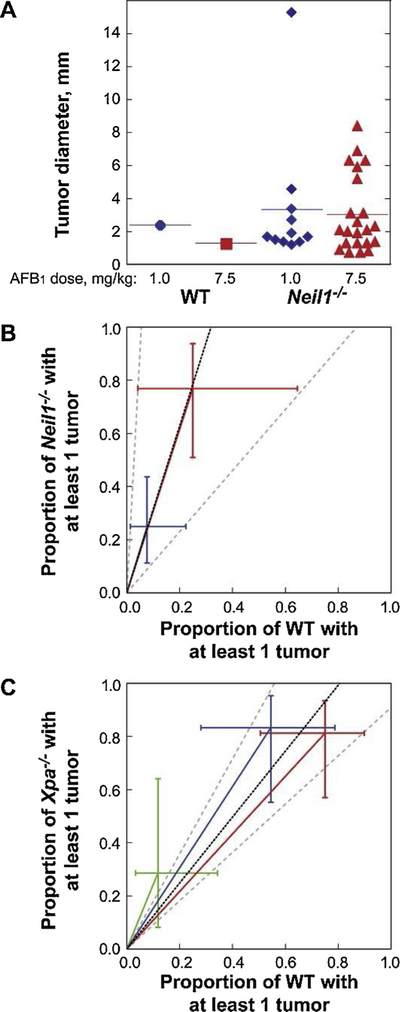
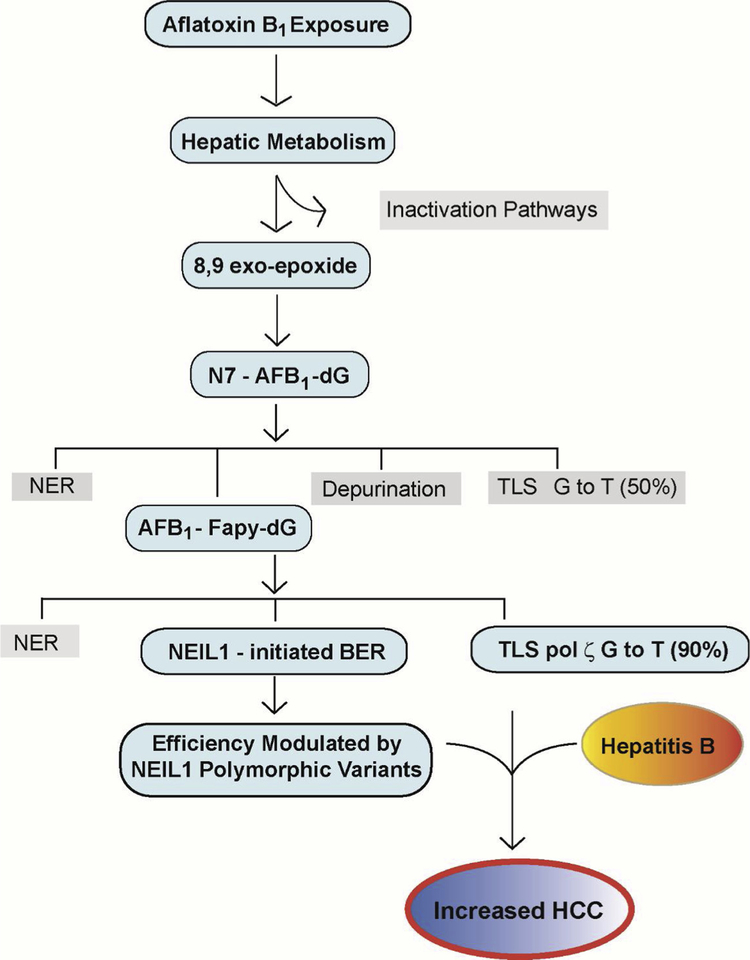
Chronic dietary exposure to aflatoxin B1 (AFB1), concomitant with hepatitis B infection is associated with a significant increased risk for hepatocellular carcinomas (HCCs) in people living in Southeast Asia and sub-Saharan Africa. Human exposures to AFB1 occur through the consumption of foods that are contaminated with pervasive molds, including Aspergillus flavus. Even though dietary exposures to aflatoxins constitute the second largest global environmental risk factor for cancer development, there are still significant questions concerning the molecular mechanisms driving carcinogenesis and what factors may modulate an individual's risk for HCC. The objective of this review is to summarize key discoveries that established the association of chronic inflammation (most commonly associated with hepatitis B viral (HBV) infection) and environmental exposures to aflatoxin with increased HCC risk. Special emphasis will be given to recent investigations that have: 1) refined the aflatoxin-associated mutagenic signature, 2) expanded the DNA repair mechanisms that limit mutagenesis via adduct removal prior to replication-induced mutagenesis, 3) implicated a specific DNA polymerase in the error-prone bypass and resulting mutagenesis, and 4) identified human polymorphic variants that may modulate individual susceptibility to aflatoxin-induced cancers. Collectively, these investigations revealed that specific sequence contexts are differentially resistant against, or prone to, aflatoxin-induced mutagenesis and that these associations are remarkably similar between in vitro and in vivo analyses. These recent investigations also established DNA polymerase ζ as the major polymerase that confers the G to T transversion signature. Additionally, although the nucleotide excision repair (NER) pathway has been previously shown to repair aflatoxin-induced DNA adducts, recent murine data demonstrated that NEIL1-initiated base excision repair was significantly more important than NER relative to the removal of the highly mutagenic AFB1-Fapy-dG adducts. These data suggest that inactivating polymorphic variants of NEIL1 could be a potential driver of HCCs in aflatoxin-exposed populations.
Keywords: DNA repair; DNA replication; Hepatocellular carcinoma; NEIL1; Polymerase zeta.
Copyright © 2019. Published by Elsevier B.V.
Conflict of interest statement
Conflict of interest
The authors declare that there are no conflicts of interest.
Figures

References
-
- Kew MC, Aflatoxins as a cause of hepatocellular carcinoma, J. Gastrointestin. Liver Dis 22 (2013) 305–310. - PubMed
-
- Sargeant K, Sheridan A, O’Kelly J, Carnaghan RBA, Toxicity associated with certain samples of groundnuts, Nature 192 (1961) 1096–1097.
-
- Lancaster MC, Jenkins FP, Philip JM, Toxicity associated with certain samples of groundnuts, Nature 192 (1961) 1095–1096.
-
- Blount WP, Turkey “x” disease, Turkeys (J. Br. Turkey Fed.) 9 (1961) 52–58.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
