

Characterization of cell fate probabilities in single-cell data with Palantir
- PMID: 30899105
- PMCID: PMC7549125
- DOI: 10.1038/s41587-019-0068-4
Characterization of cell fate probabilities in single-cell data with Palantir
Erratum in
-
Author Correction: Characterization of cell fate probabilities in single-cell data with Palantir.Nat Biotechnol. 2019 Oct;37(10):1237. doi: 10.1038/s41587-019-0282-0. Nat Biotechnol. 2019. PMID: 31534198
Abstract
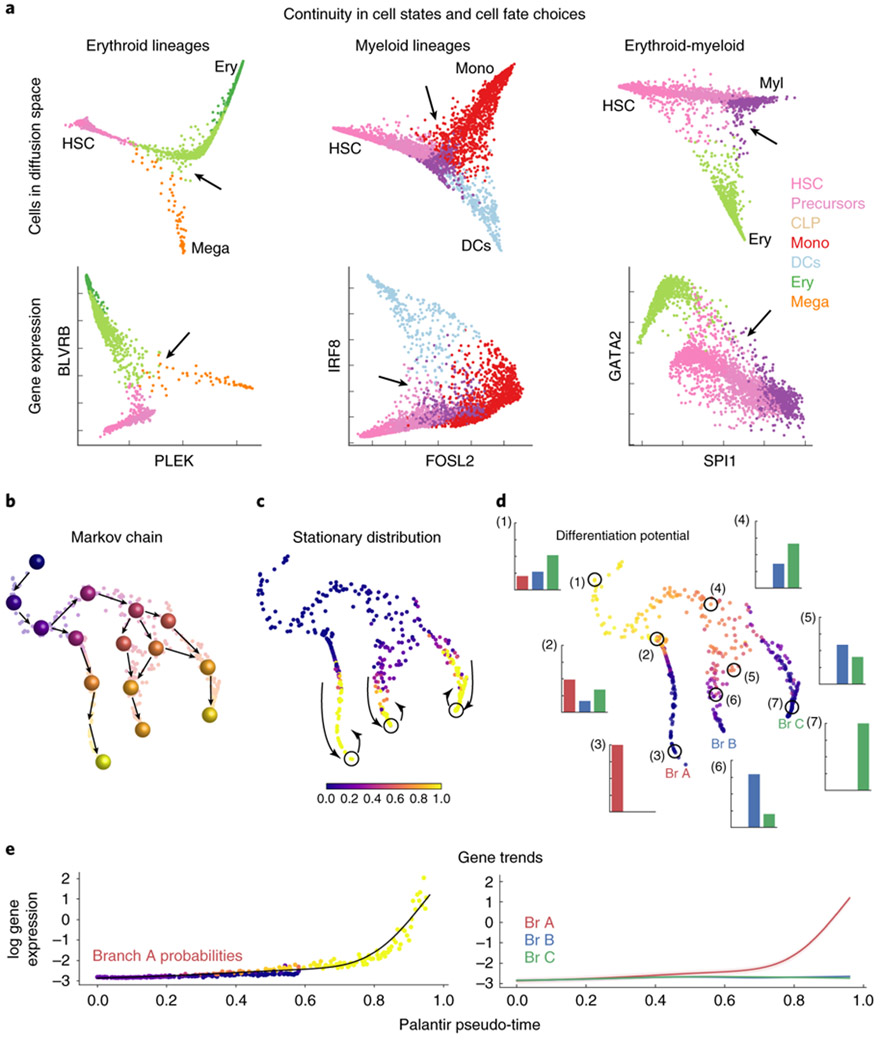
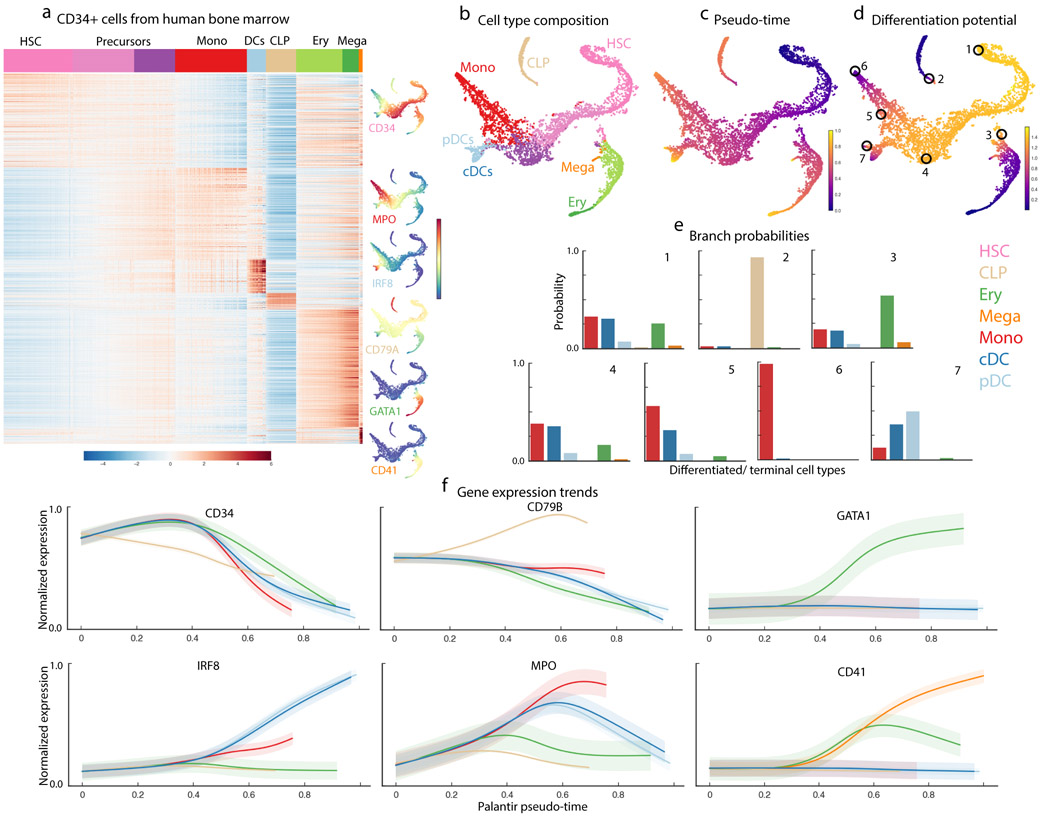
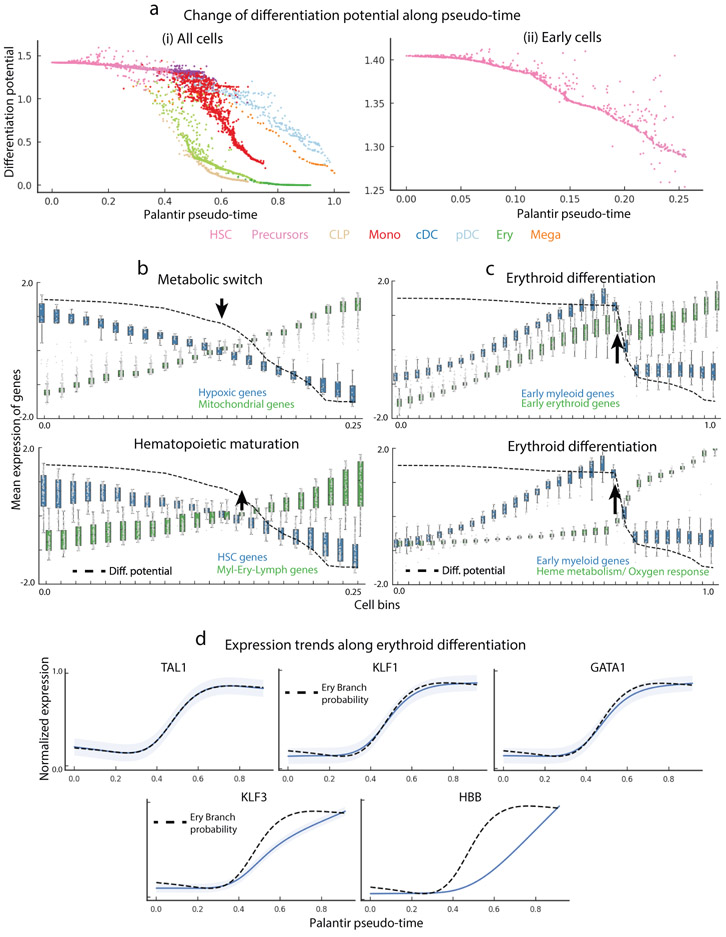
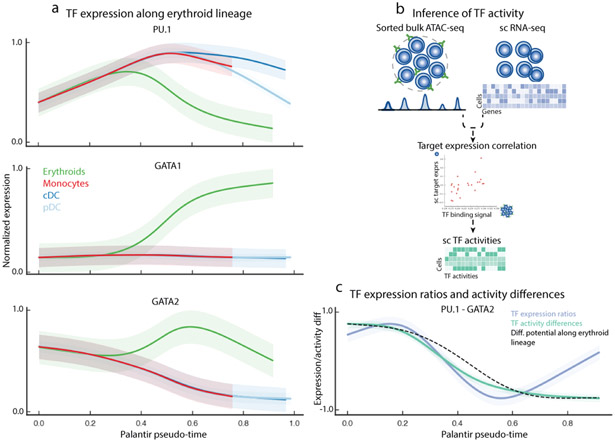
Single-cell RNA sequencing studies of differentiating systems have raised fundamental questions regarding the discrete versus continuous nature of both differentiation and cell fate. Here we present Palantir, an algorithm that models trajectories of differentiating cells by treating cell fate as a probabilistic process and leverages entropy to measure cell plasticity along the trajectory. Palantir generates a high-resolution pseudo-time ordering of cells and, for each cell state, assigns a probability of differentiating into each terminal state. We apply our algorithm to human bone marrow single-cell RNA sequencing data and detect important landmarks of hematopoietic differentiation. Palantir's resolution enables the identification of key transcription factors that drive lineage fate choice and closely track when cells lose plasticity. We show that Palantir outperforms existing algorithms in identifying cell lineages and recapitulating gene expression trends during differentiation, is generalizable to diverse tissue types, and is well-suited to resolving less-studied differentiating systems.
Figures
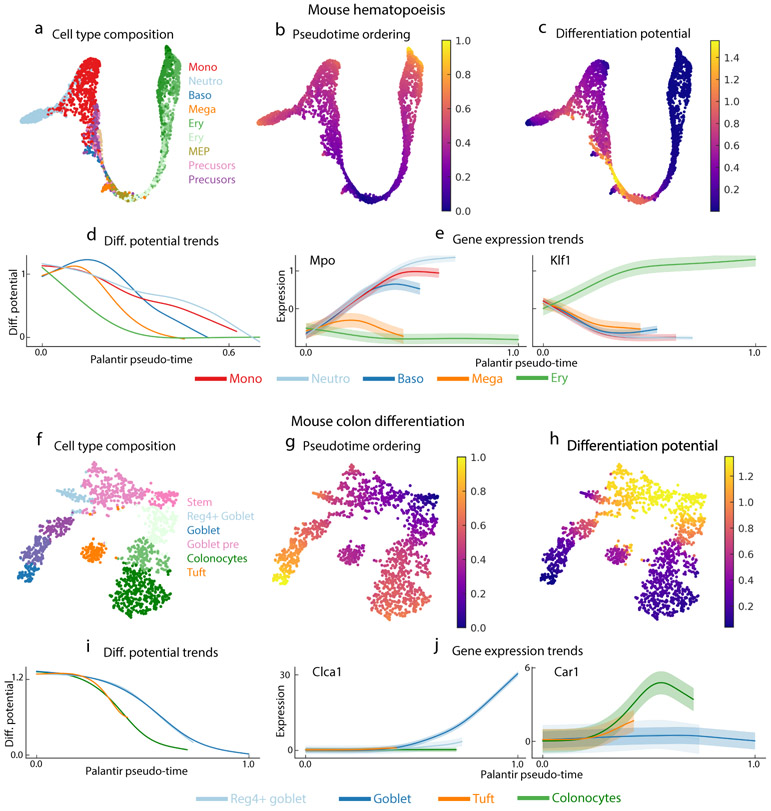
References
-
- Haghverdi L, Buttner M, Wolf FA, Buettner F & Theis FJ Diffusion pseudotime robustly reconstructs lineage branching. Nature methods 13, 845–848 (2016). - PubMed
Online Methods References
-
- Buettner F et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nature biotechnology 33, 155–160 (2015). - PubMed
-
- van der Maaten LPJ & Hinton GE Visualizing High-Dimensional Data Using t-SNE. Journal of Machine Learning Researc 9, 2579–2605 (2008).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
