

Depletion of H3K79 methyltransferase Dot1L promotes cell invasion and cancer stem-like cell property in ovarian cancer
- PMID: 30899413
- PMCID: PMC6413254
Depletion of H3K79 methyltransferase Dot1L promotes cell invasion and cancer stem-like cell property in ovarian cancer
Abstract
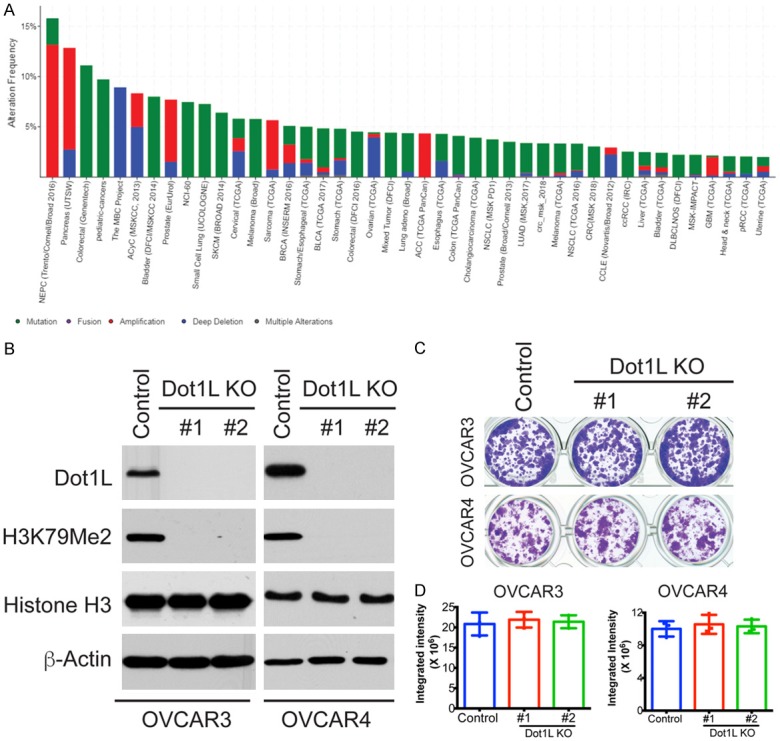
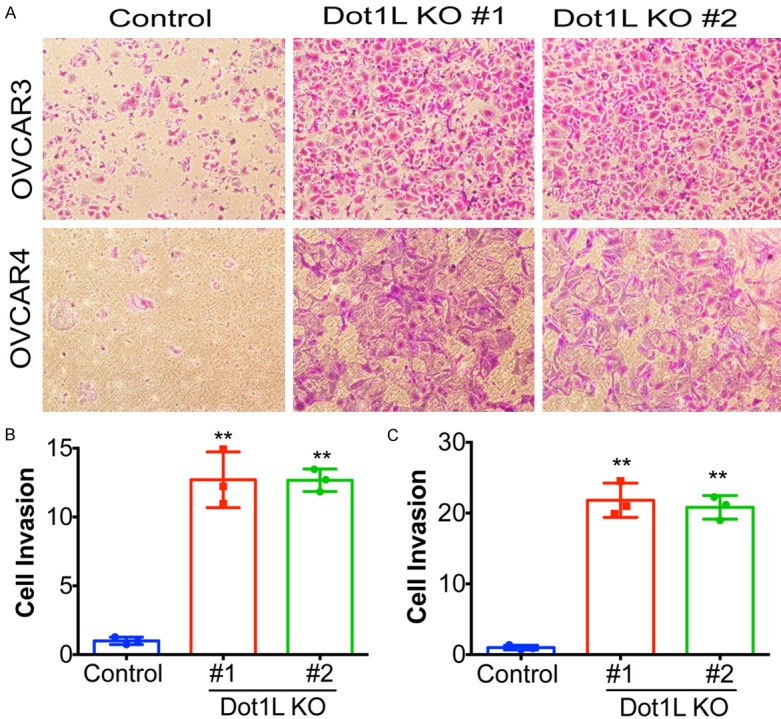
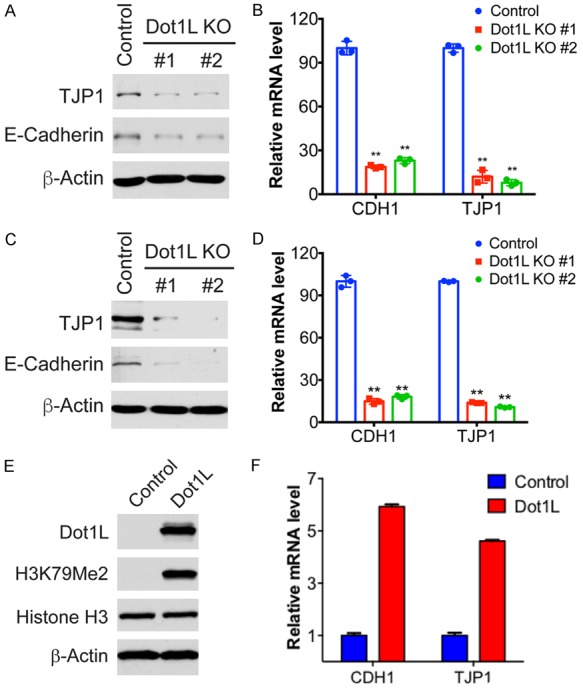
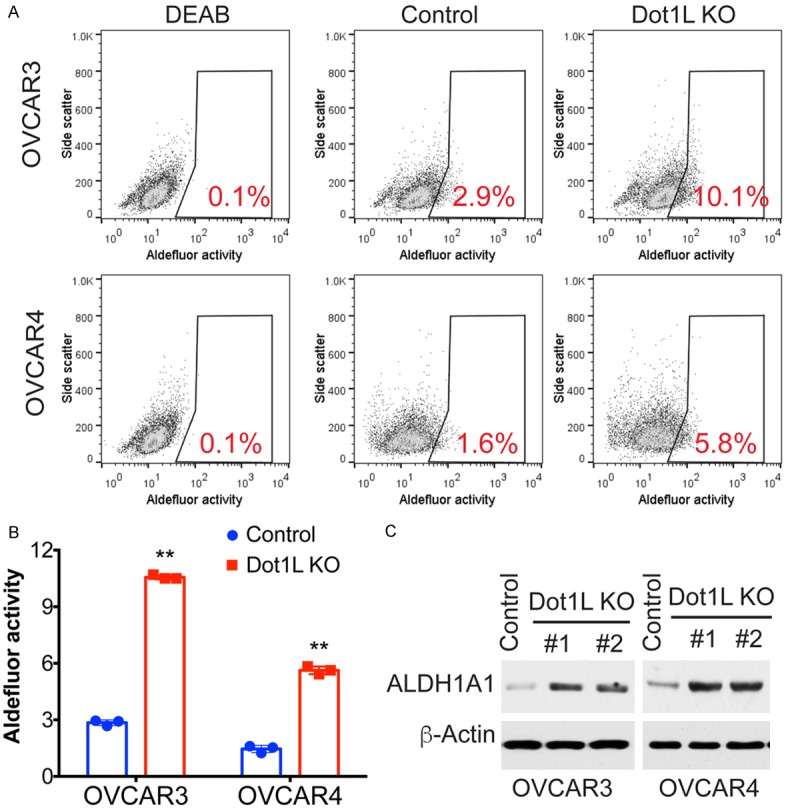
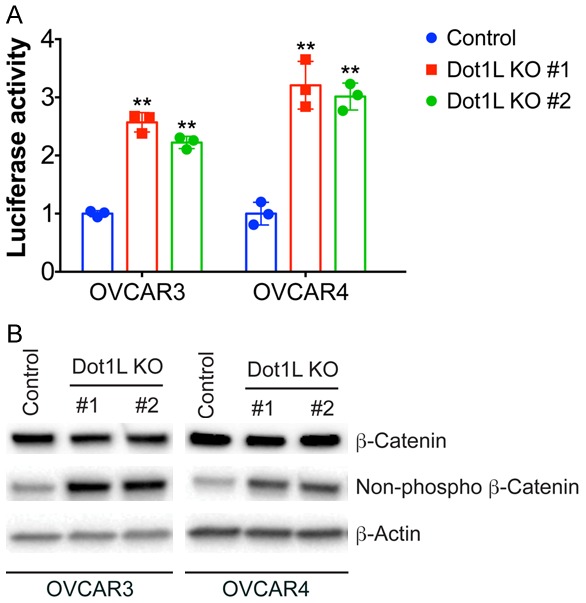
DOT1-like protein (Dot1L) is the sole methyltransferase for methylation of lysine 79 in histone H3. Dot1L-dependent H3K79 methylation is involved in many biological processes, including telomeric silencing, cell cycle regulation, transcriptional activation and DNA repair. Genome-wide sequencing studies have revealed recurrent deletion and mutations of Dot1L gene in many types of human malignancies including ovarian cancer, however the role of Dot1L in ovarian cancer are largely unknown. To demonstrate the role of Dot1L in ovarian cancer, the expression of Dot1L was knocked out in ovarian cancer cells using CRISPR/Cas9 technology in the present study. Dot1L loss showed minimal effect on cell growth, but significantly promoted cell invasion and induced cancer stem-like cell property in ovarian cancer cells. Mechanistically, loss of Dot1L downregulated the expression of tight junction makers E-Cadherin and TJP1 and upregulated the expression of ALDH1A1 through Wnt signaling activation. Our data indicate potential tumor suppressor function of Dot1L in ovarian cancer, which is correlated with observed deletion of Dot1L gene in ovarian cancer patients, further study is granted to elucidate the function of Dot1L in tumorigenesis and progression in ovarian cancer.
Keywords: DOT1-like protein; Wnt signaling; cancer stem cell; cell invasion; ovarian cancer.
Conflict of interest statement
None.
Figures
Similar articles
-
DOT1 L Regulates Ovarian Cancer Stem Cells by Activating β-catenin Signaling.Mol Cancer Res. 2023 Feb 1;21(2):140-154. doi: 10.1158/1541-7786.MCR-22-0418. Mol Cancer Res. 2023. PMID: 36318113 Free PMC article.
-
Prognostic and therapeutic value of disruptor of telomeric silencing-1-like (DOT1L) expression in patients with ovarian cancer.J Hematol Oncol. 2017 Jan 23;10(1):29. doi: 10.1186/s13045-017-0400-8. J Hematol Oncol. 2017. PMID: 28114995 Free PMC article.
-
The histone methyltransferase Dot1/DOT1L as a critical regulator of the cell cycle.Cell Cycle. 2014;13(5):726-38. doi: 10.4161/cc.28104. Epub 2014 Feb 6. Cell Cycle. 2014. PMID: 24526115 Free PMC article. Review.
-
Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation.J Biol Chem. 2012 Feb 17;287(8):5588-99. doi: 10.1074/jbc.M111.328138. Epub 2011 Dec 21. J Biol Chem. 2012. PMID: 22190683 Free PMC article.
-
The diverse functions of Dot1 and H3K79 methylation.Genes Dev. 2011 Jul 1;25(13):1345-58. doi: 10.1101/gad.2057811. Genes Dev. 2011. PMID: 21724828 Free PMC article. Review.
Cited by
-
DOT1 L Regulates Ovarian Cancer Stem Cells by Activating β-catenin Signaling.Mol Cancer Res. 2023 Feb 1;21(2):140-154. doi: 10.1158/1541-7786.MCR-22-0418. Mol Cancer Res. 2023. PMID: 36318113 Free PMC article.
-
Comprehensive analysis of histone methylation modification regulators for predicting prognosis and drug sensitivity in lung adenocarcinoma.Front Cell Dev Biol. 2022 Oct 3;10:991980. doi: 10.3389/fcell.2022.991980. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 36263018 Free PMC article.
-
Reprogramming: identifying the mechanisms that safeguard cell identity.Development. 2019 Dec 2;146(23):dev182170. doi: 10.1242/dev.182170. Development. 2019. PMID: 31792064 Free PMC article. Review.
-
The roles of histone modifications in tumorigenesis and associated inhibitors in cancer therapy.J Natl Cancer Cent. 2022 Sep 28;2(4):277-290. doi: 10.1016/j.jncc.2022.09.002. eCollection 2022 Dec. J Natl Cancer Cent. 2022. PMID: 39036551 Free PMC article. Review.
-
Silencing or inhibition of H3K79 methyltransferase DOT1L induces cell cycle arrest by epigenetically modulating c-Myc expression in colorectal cancer.Clin Epigenetics. 2019 Dec 30;11(1):199. doi: 10.1186/s13148-019-0778-y. Clin Epigenetics. 2019. PMID: 31888761 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous