

Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders
- PMID: 30901538
- PMCID: PMC6432948
- DOI: 10.1016/j.cell.2019.01.015
Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders
Abstract
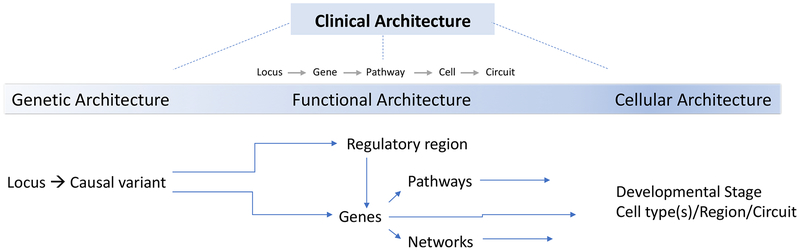
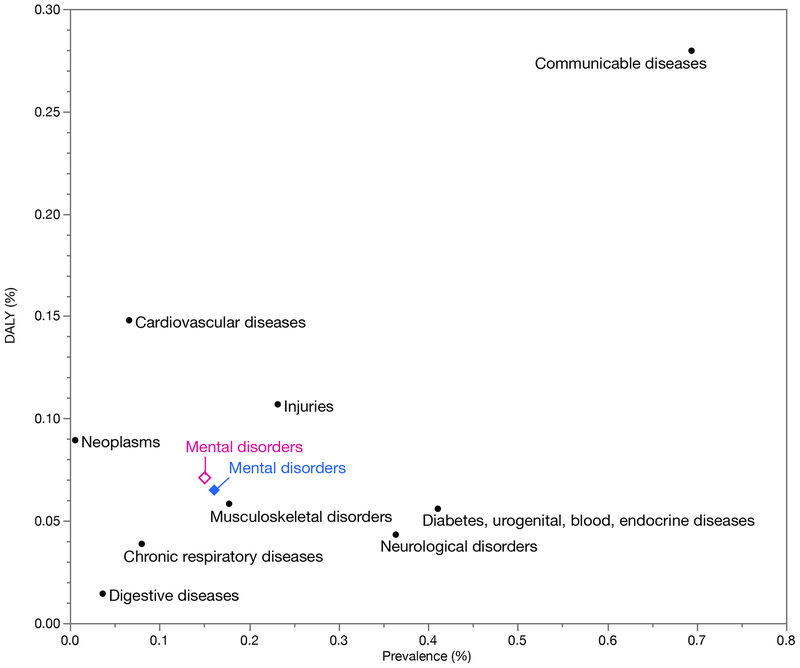
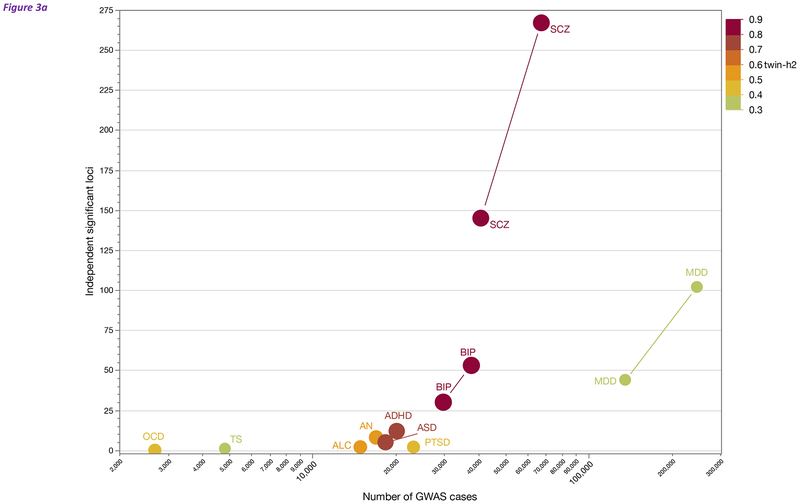
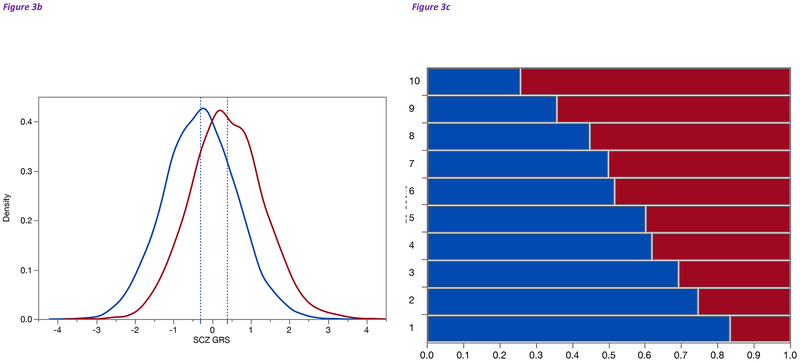
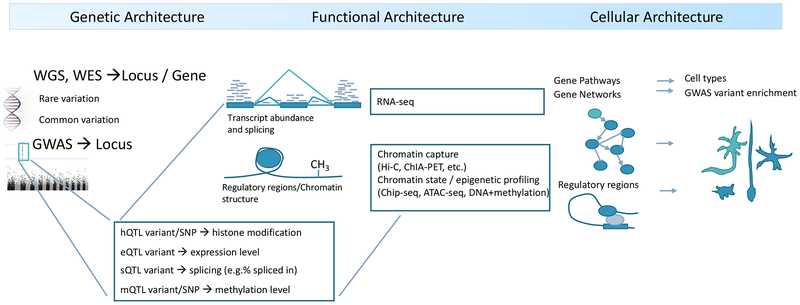
Studies of the genetics of psychiatric disorders have become one of the most exciting and fast-moving areas in human genetics. A decade ago, there were few reproducible findings, and now there are hundreds. In this review, we focus on the findings that have illuminated the genetic architecture of psychiatric disorders and the challenges of using these findings to inform our understanding of pathophysiology. The evidence is now overwhelming that psychiatric disorders are "polygenic"-that many genetic loci contribute to risk. With the exception of a subset of those with ASD, few individuals with a psychiatric disorder have a single, deterministic genetic cause; rather, developing a psychiatric disorder is influenced by hundreds of different genetic variants, consistent with a polygenic model. As progressively larger studies have uncovered more about their genetic architecture, the need to elucidate additional architectures has become clear. Even if we were to have complete knowledge of the genetic architecture of a psychiatric disorder, full understanding requires deep knowledge of the functional genomic architecture-the implicated loci impact regulatory processes that influence gene expression and the functional coordination of genes that control biological processes. Following from this is cellular architecture: of all brain regions, cell types, and developmental stages, where and when are the functional architectures operative? Given that the genetic architectures of different psychiatric disorders often strongly overlap, we are challenged to re-evaluate and refine the diagnostic architectures of psychiatric disorders using fundamental genetic and neurobiological data.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts
PF Sullivan reports the following potentially competing financial interests. Current: Lundbeck (advisory committee, grant recipient). Past three years: Pfizer (scientific advisory board), Element Genomics (consultation fee), and Roche (speaker reimbursement). DH Geschwind has the following disclosures: Research funding from Takeda pharmaceuticals, and serving as a scientific advisor for Falcon Computing, Ovid Therapeutics, Axial Biosciences, Acurastem, and Third Rock Ventures.
Figures
References
-
- Arnold CD, Gerlach D, Stelzer C, Boryn LM, Rath M, and Stark A (2013). Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science 339, 1074–1077. - PubMed
-
- Billstedt E, Gillberg IC, and Gillberg C (2007). Autism in adults: symptom patterns and early childhood predictors. Use of the DISCO in a community sample followed from childhood. J Child Psychol Psychiatry 48, 1102–1110. - PubMed
Publication types
MeSH terms
Grants and funding
- R01 MH094714/MH/NIMH NIH HHS/United States
- P50 MH106438/MH/NIMH NIH HHS/United States
- R01 HD065280/HD/NICHD NIH HHS/United States
- R01 MH077139/MH/NIMH NIH HHS/United States
- R01 MH100027/MH/NIMH NIH HHS/United States
- R01 MH100028/MH/NIMH NIH HHS/United States
- U01 MH109536/MH/NIMH NIH HHS/United States
- P50 HD055784/HD/NICHD NIH HHS/United States
- U01 MH115746/MH/NIMH NIH HHS/United States
- R01 MH081754/MH/NIMH NIH HHS/United States
- R01 MH100900/MH/NIMH NIH HHS/United States
- U01 MH109532/MH/NIMH NIH HHS/United States
- R33 MH087898/MH/NIMH NIH HHS/United States
- R01 MH109912/MH/NIMH NIH HHS/United States
- U01 MH109528/MH/NIMH NIH HHS/United States
- R01 NS073871/NS/NINDS NIH HHS/United States
- U01 MH105991/MH/NIMH NIH HHS/United States
- R01 MH110927/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
