

Global Genetic Networks and the Genotype-to-Phenotype Relationship
- PMID: 30901552
- PMCID: PMC6817365
- DOI: 10.1016/j.cell.2019.01.033
Global Genetic Networks and the Genotype-to-Phenotype Relationship
Abstract
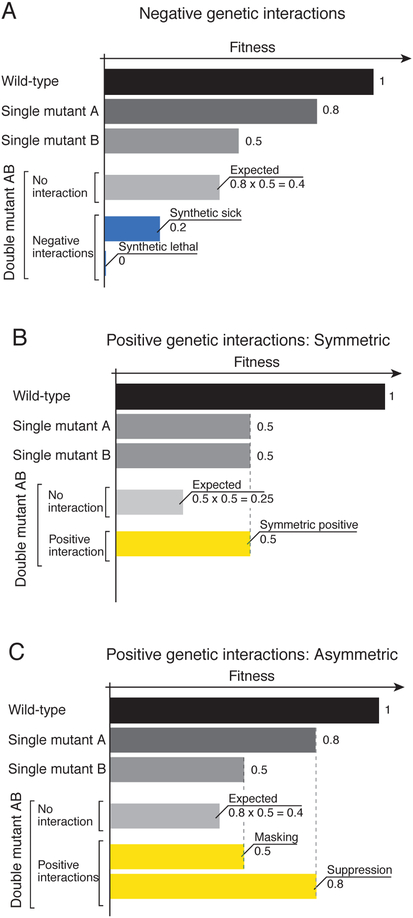
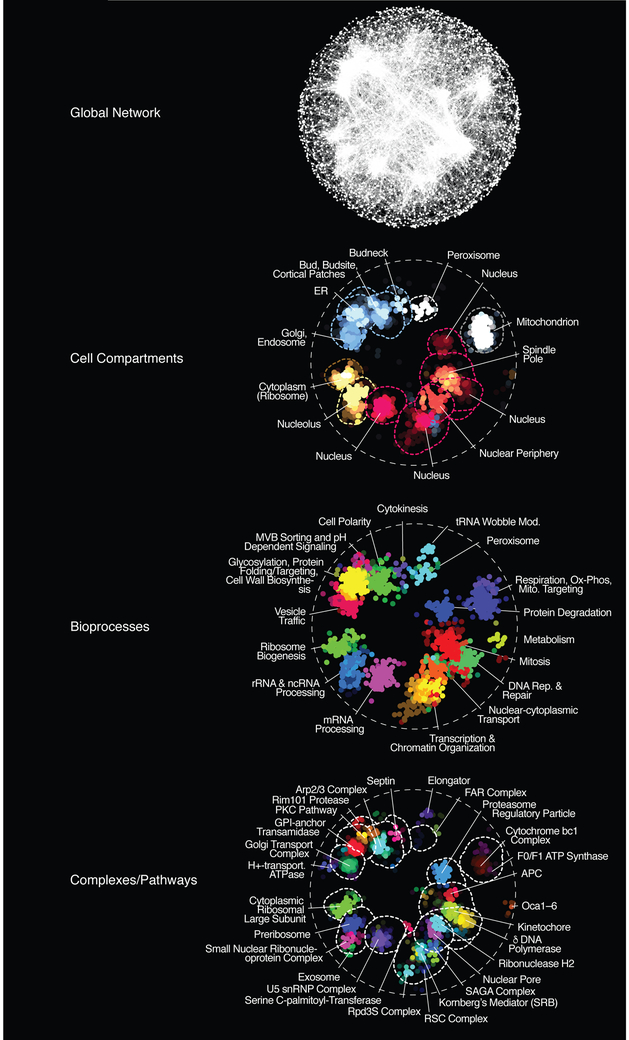
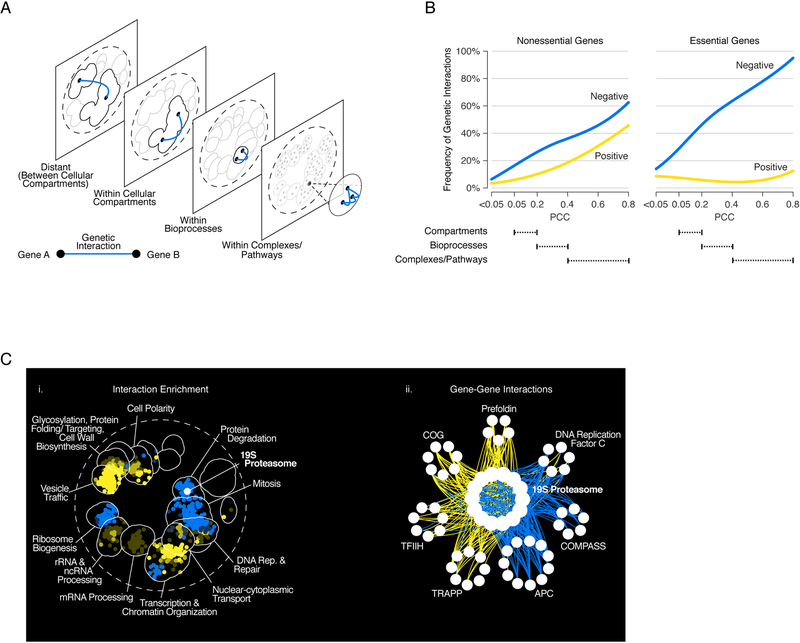
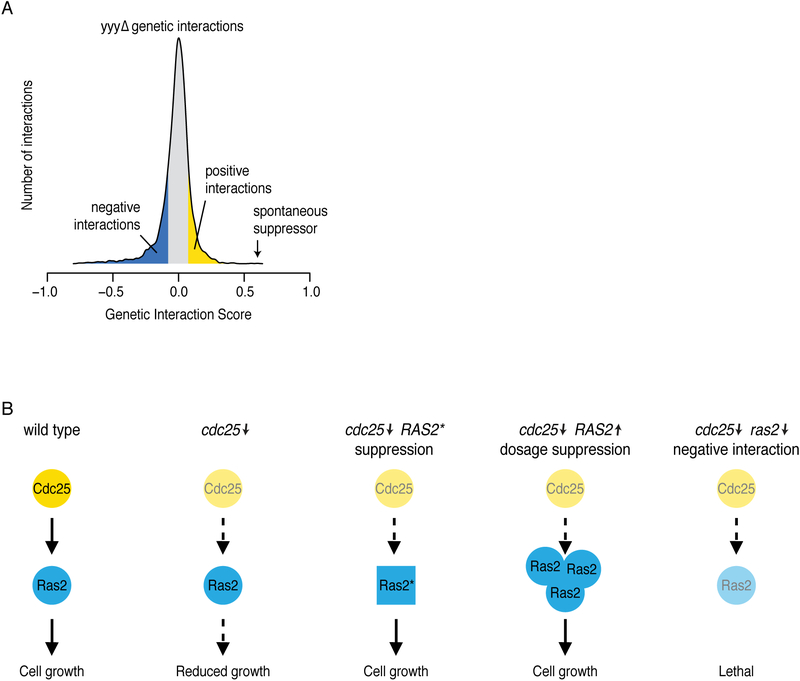
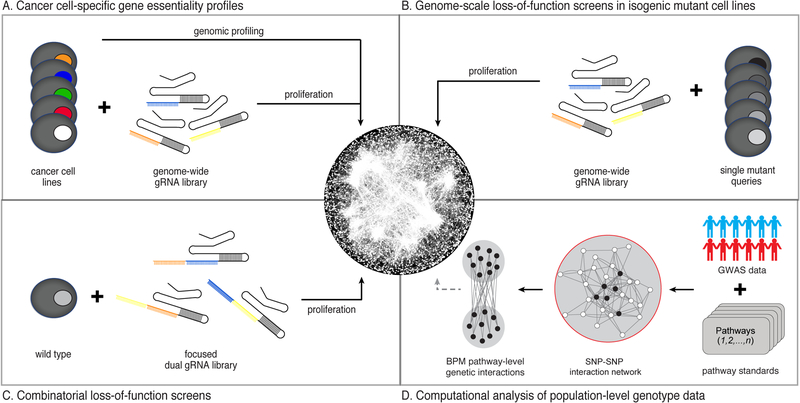
Genetic interactions identify combinations of genetic variants that impinge on phenotype. With whole-genome sequence information available for thousands of individuals within a species, a major outstanding issue concerns the interpretation of allelic combinations of genes underlying inherited traits. In this Review, we discuss how large-scale analyses in model systems have illuminated the general principles and phenotypic impact of genetic interactions. We focus on studies in budding yeast, including the mapping of a global genetic network. We emphasize how information gained from work in yeast translates to other systems, and how a global genetic network not only annotates gene function but also provides new insights into the genotype-to-phenotype relationship.
Copyright © 2019. Published by Elsevier Inc.
Figures
References
-
- Agarwala A, and Fisher DS (2018). Adaptive walks on high-dimensional fitness landscapes and seascapes with distance-dependent statistics. bioRxiv. - PubMed
-
- Ashworth A, and Lord CJ (2018). Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nature Reviews Clinical Oncology 15, 564–576. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
