

The past, present and future of anti-malarial medicines
- PMID: 30902052
- PMCID: PMC6431062
- DOI: 10.1186/s12936-019-2724-z
The past, present and future of anti-malarial medicines
Abstract
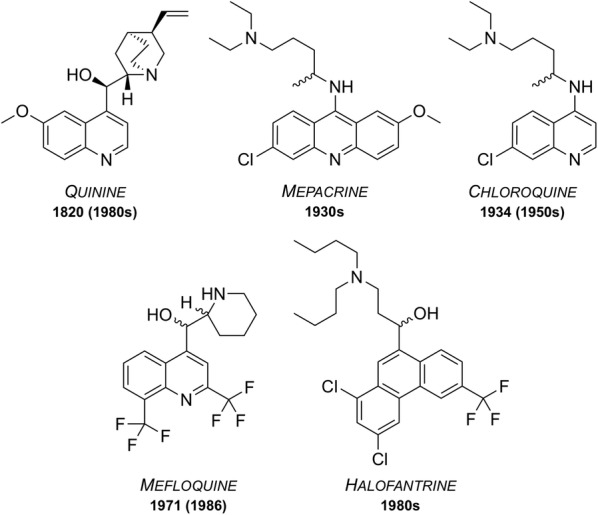
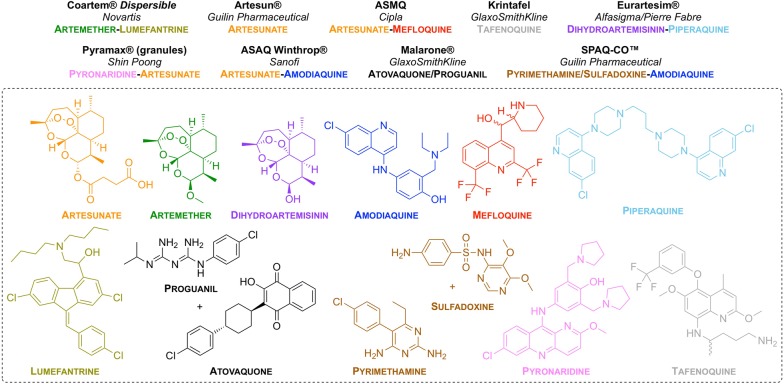
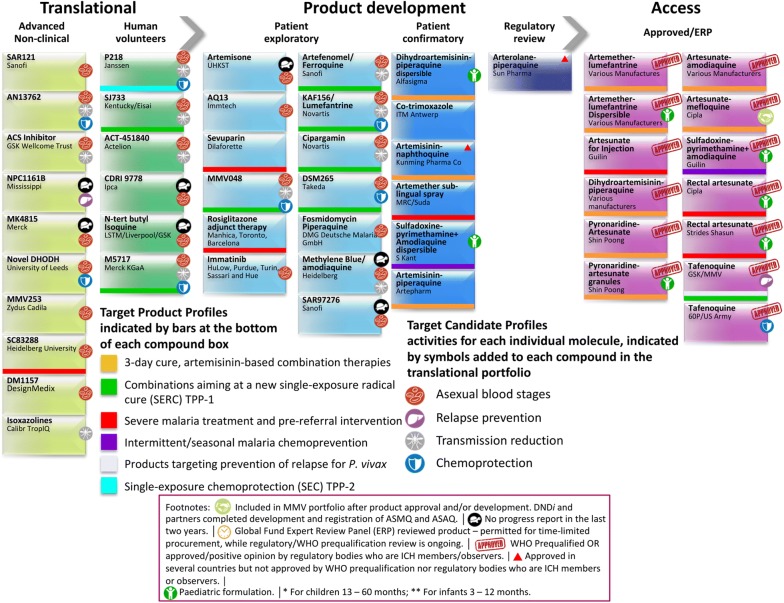
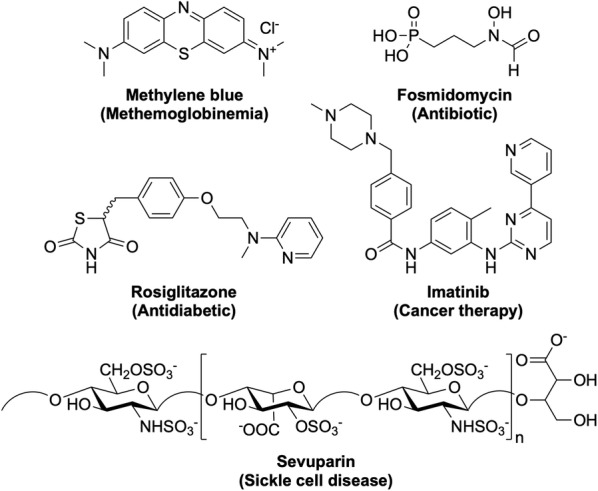
Great progress has been made in recent years to reduce the high level of suffering caused by malaria worldwide. Notably, the use of insecticide-treated mosquito nets for malaria prevention and the use of artemisinin-based combination therapy (ACT) for malaria treatment have made a significant impact. Nevertheless, the development of resistance to the past and present anti-malarial drugs highlights the need for continued research to stay one step ahead. New drugs are needed, particularly those with new mechanisms of action. Here the range of anti-malarial medicines developed over the years are reviewed, beginning with the discovery of quinine in the early 1800s, through to modern day ACT and the recently-approved tafenoquine. A number of new potential anti-malarial drugs currently in development are outlined, along with a description of the hit to lead campaign from which it originated. Finally, promising novel mechanisms of action for these and future anti-malarial medicines are outlined.
Keywords: Drug development; Drug discovery; Malaria; Mechanism of action; Plasmodium.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
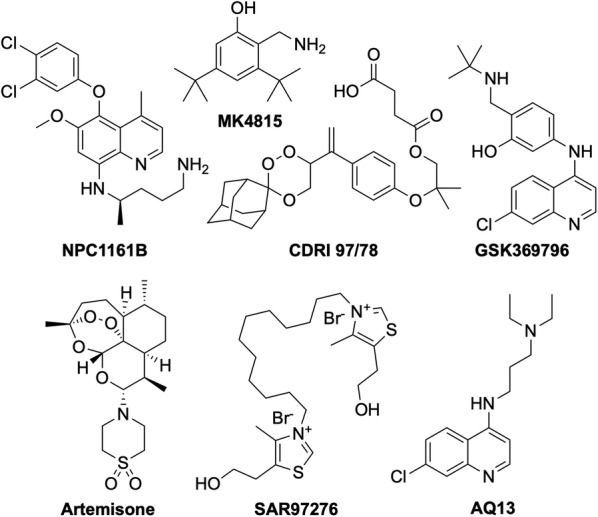
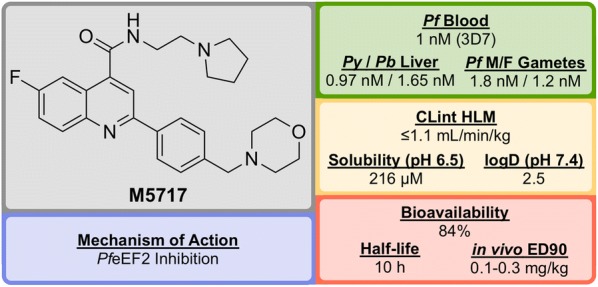
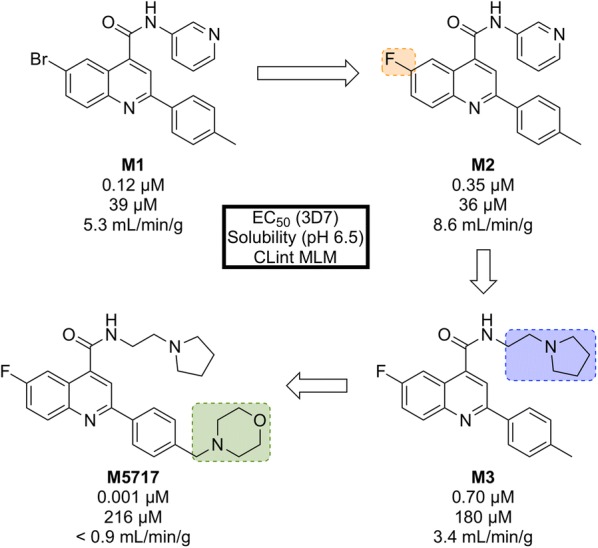
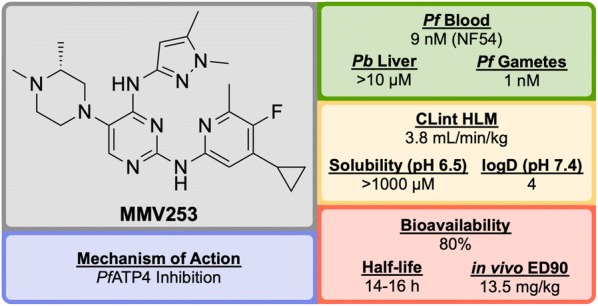
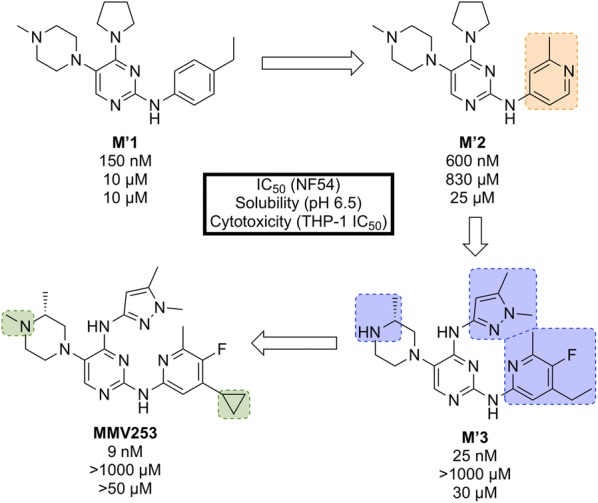
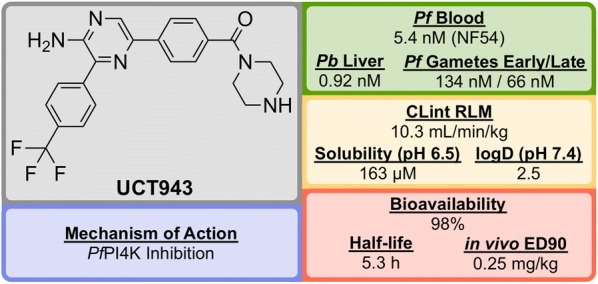
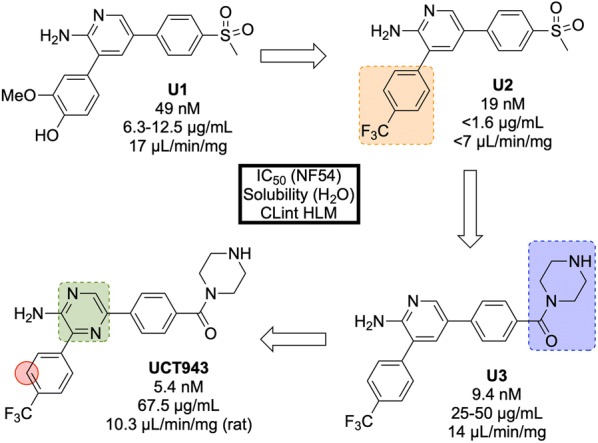
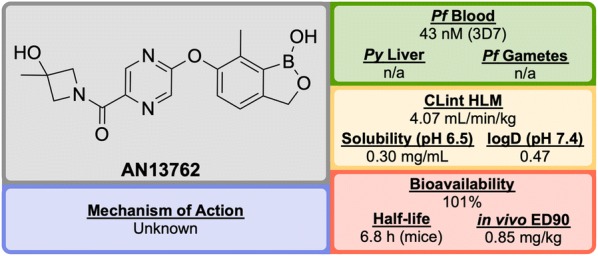
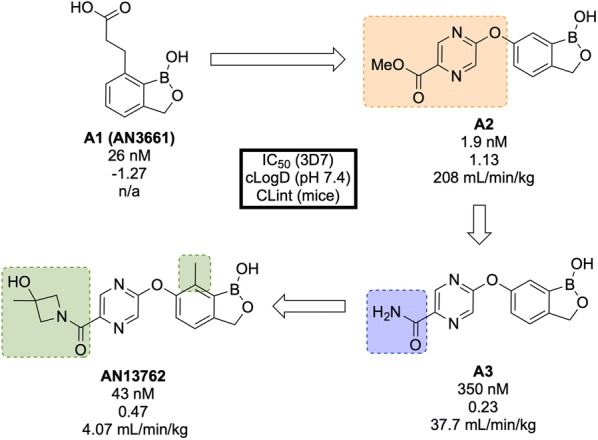
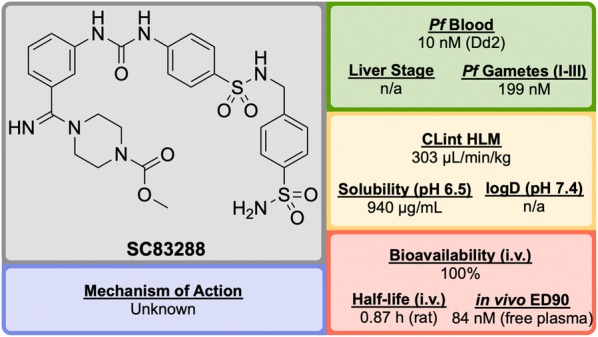
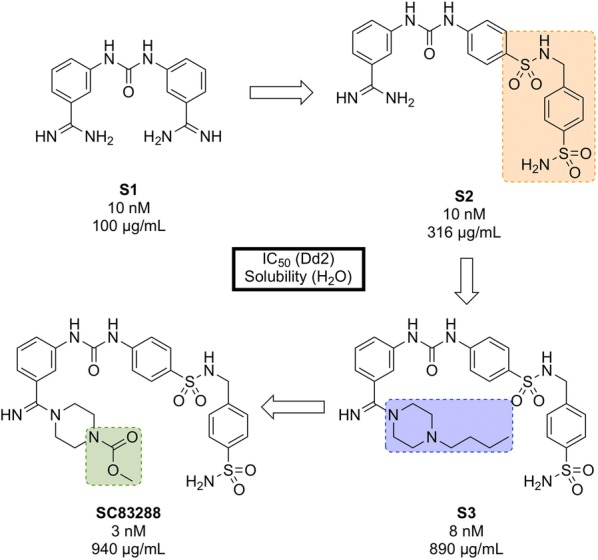
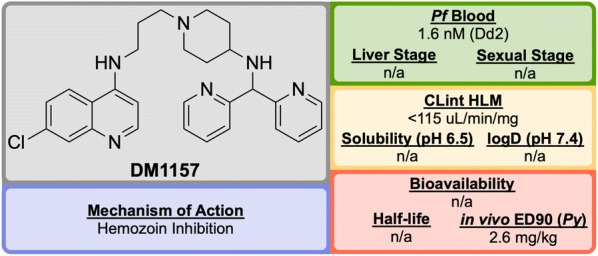
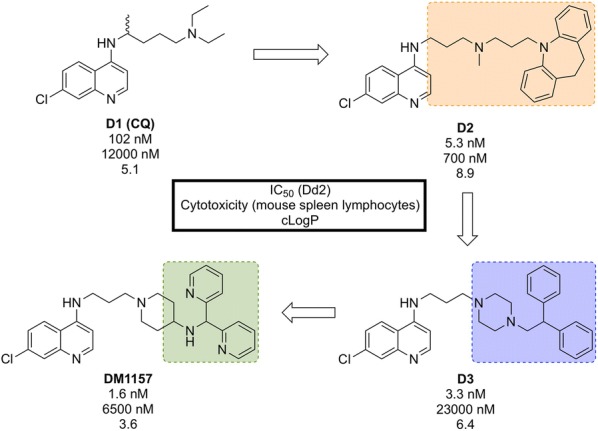
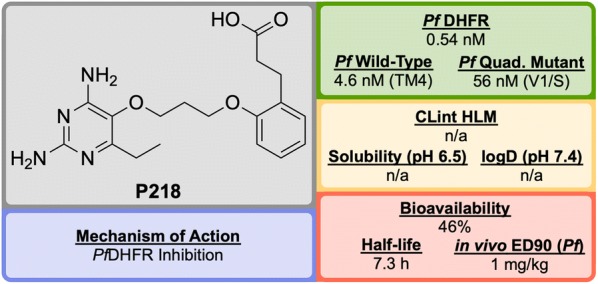
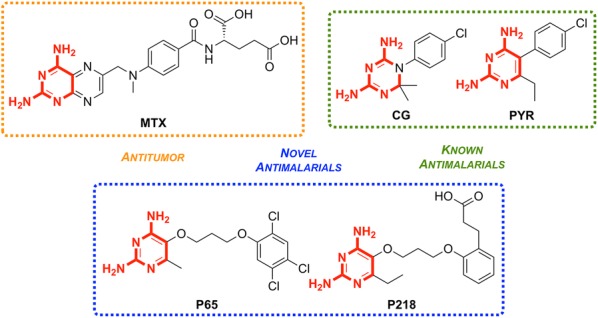
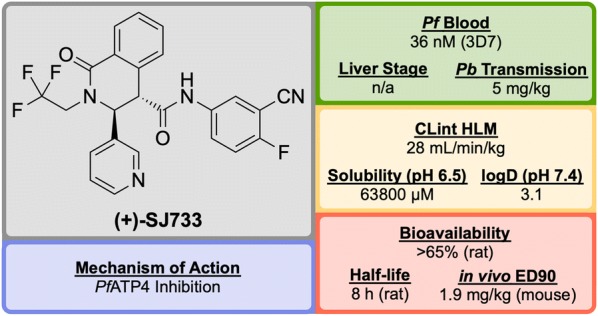
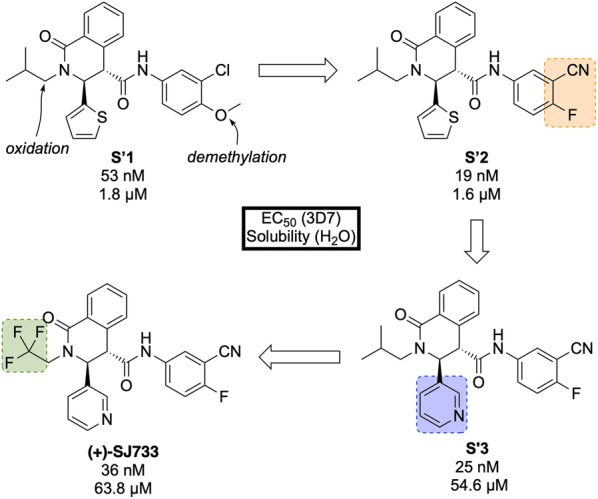
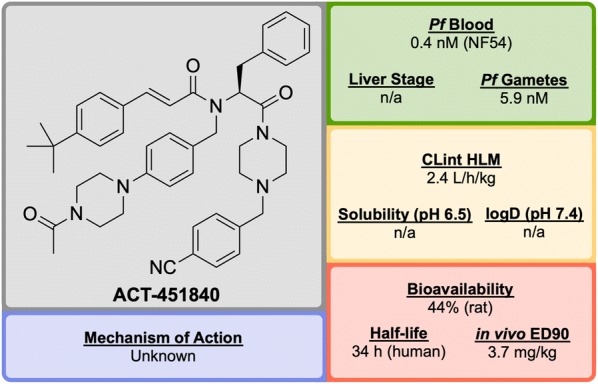
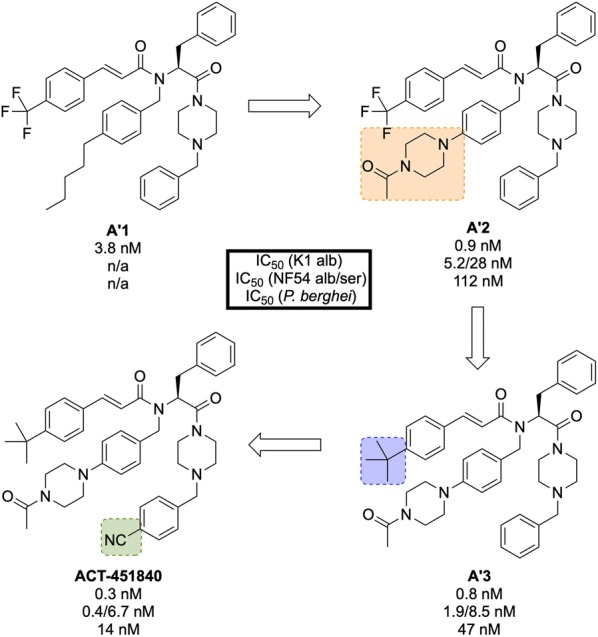
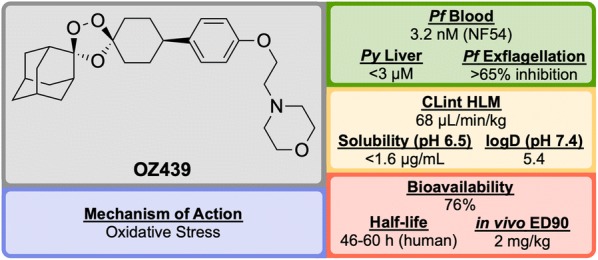
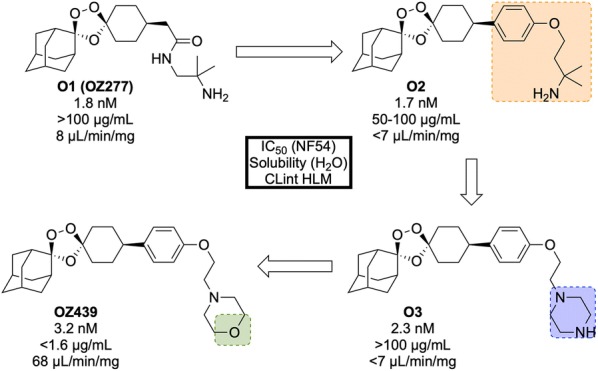
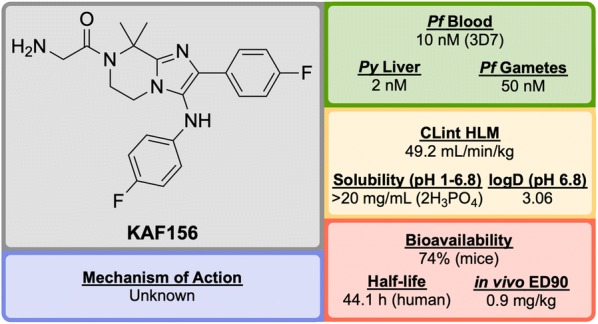
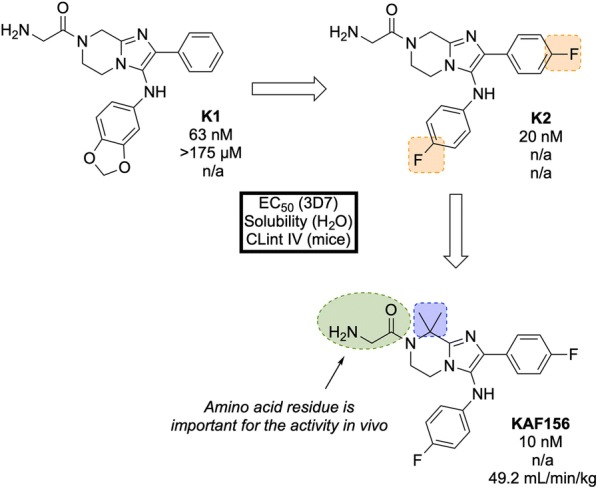
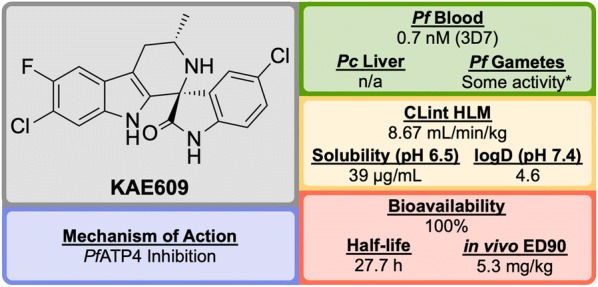
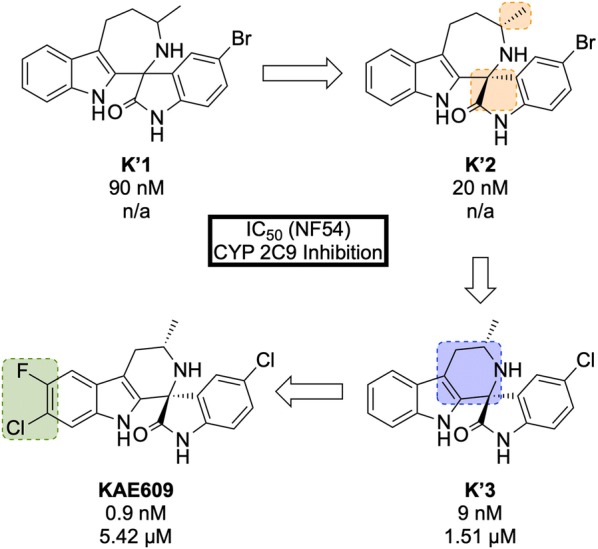
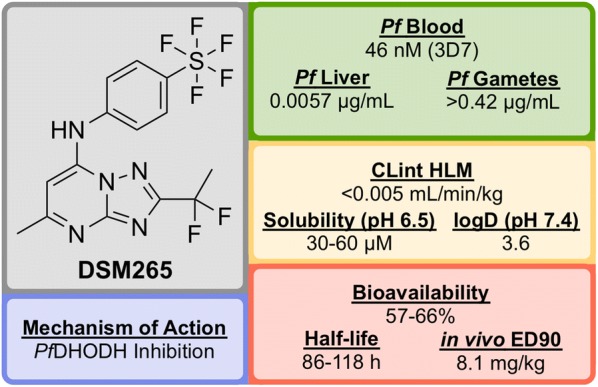
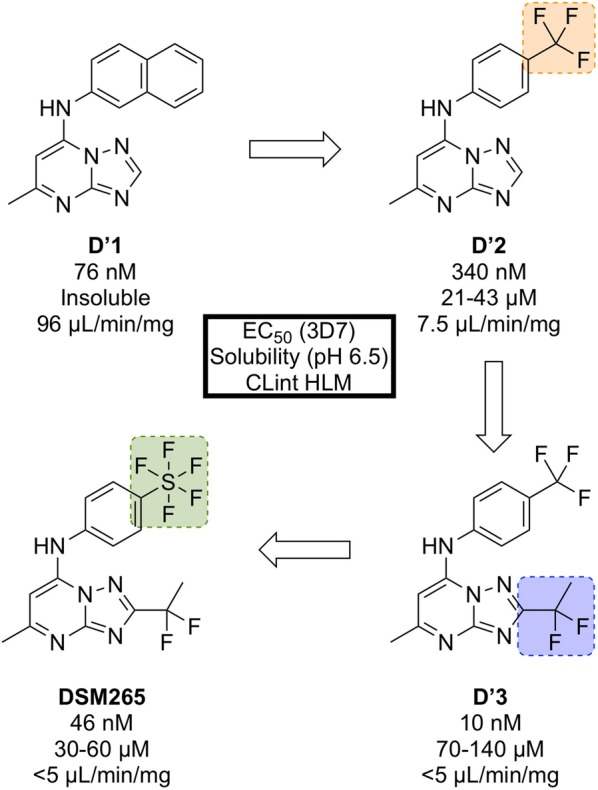
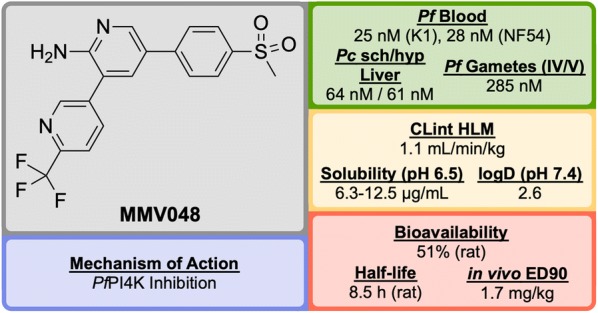
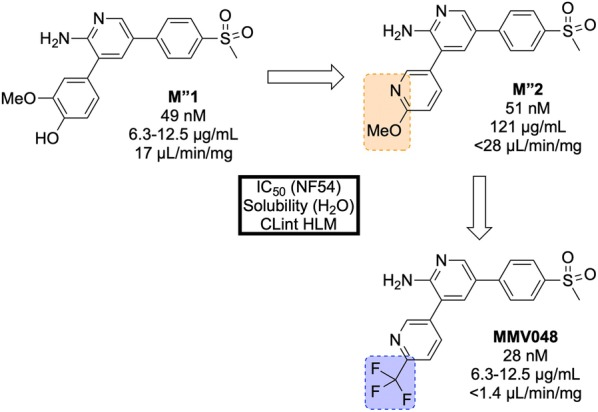
References
-
- World Malaria Report 2018. https://www.who.int/malaria/publications/world-malaria-report-2018/en/. Accessed 27 Nov 2018.
-
- Cheney C. Fight against malaria stalling and could reverse, warns 2017 World Malaria Report. http://theconversation.com/global-malaria-report-reveals-africas-hits-an.... Accessed 24 Mar 2018.
-
- Mwangi T. Global malaria report reveals Africa’s hits and missed: here’s what to do. https://www.devex.com/news/fight-against-malaria-stalling-and-could-reve.... Accessed 24 Mar 2018.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
