

X-linked myotubular myopathy: A prospective international natural history study
- PMID: 30902907
- PMCID: PMC6550499
- DOI: 10.1212/WNL.0000000000007319
X-linked myotubular myopathy: A prospective international natural history study
Abstract
Objectives: Because X-linked myotubular myopathy (XLMTM) is a rare neuromuscular disease caused by mutations in the MTM1 gene with a large phenotypic heterogeneity, to ensure clinical trial readiness, it was mandatory to better quantify disease burden and determine best outcome measures.
Methods: We designed an international prospective and longitudinal natural history study in patients with XLMTM and assessed muscle strength and motor and respiratory functions over the first year of follow-up. The humoral immunity against adeno-associated virus serotype 8 was also monitored.
Results: Forty-five male patients aged 3.5 months to 56.8 years were enrolled between May 2014 and May 2017. Thirteen patients had a mild phenotype (no ventilation support), 7 had an intermediate phenotype (ventilation support less than 12 hours a day), and 25 had a severe phenotype (ventilation support 12 or more hours a day). Most strength and motor function assessments could be performed even in very weak patients. Motor Function Measure 32 total score, grip and pinch strengths, and forced vital capacity, forced expiratory volume in the first second of exhalation, and peak cough flow measures discriminated the 3 groups of patients. Disease history revealed motor milestone loss in several patients. Longitudinal data on 37 patients showed that the Motor Function Measure 32 total score significantly decreased by 2%. Of the 38 patients evaluated, anti-adeno-associated virus type 8 neutralizing activity was detected in 26% with 2 patients having an inhibitory titer >1:10.
Conclusions: Our data confirm that XLMTM is slowly progressive for male survivors regardless of their phenotype and provide outcome validation and natural history data that can support clinical development in this population.
Clinicaltrialsgov identifier: NCT02057705.
© 2019 American Academy of Neurology.
Figures
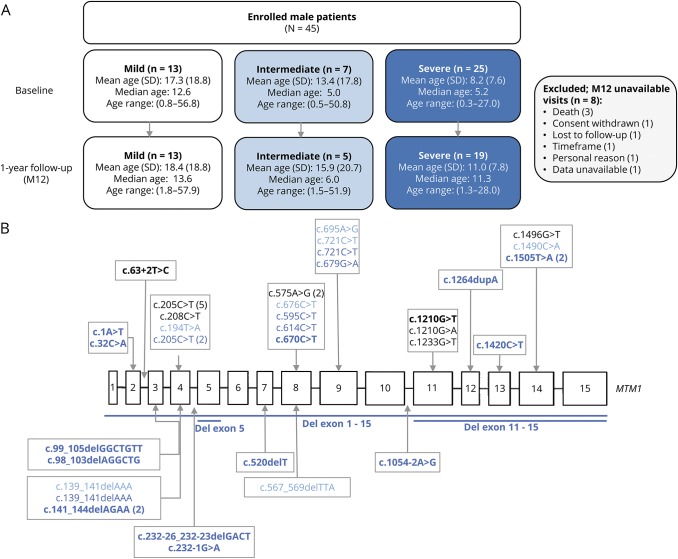
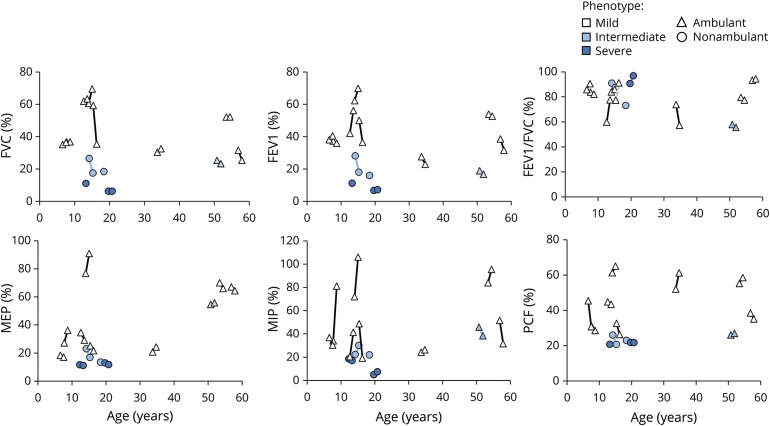
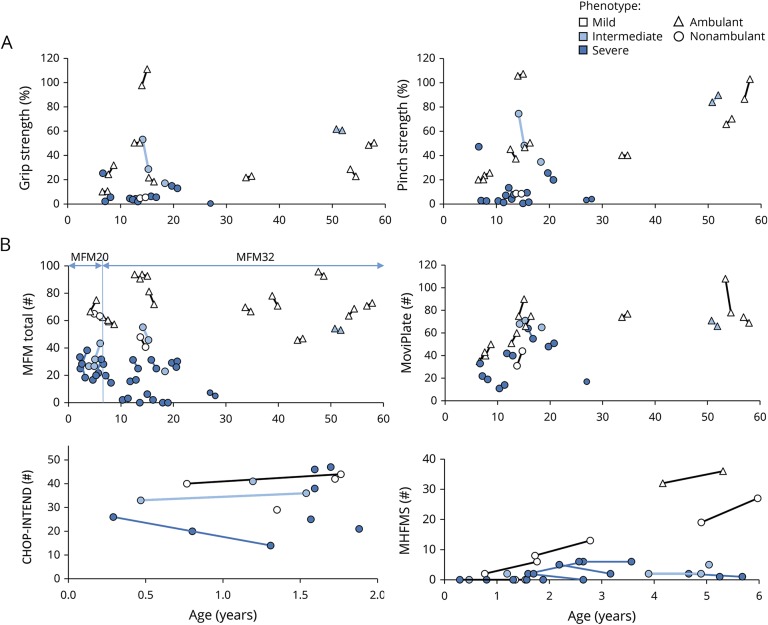
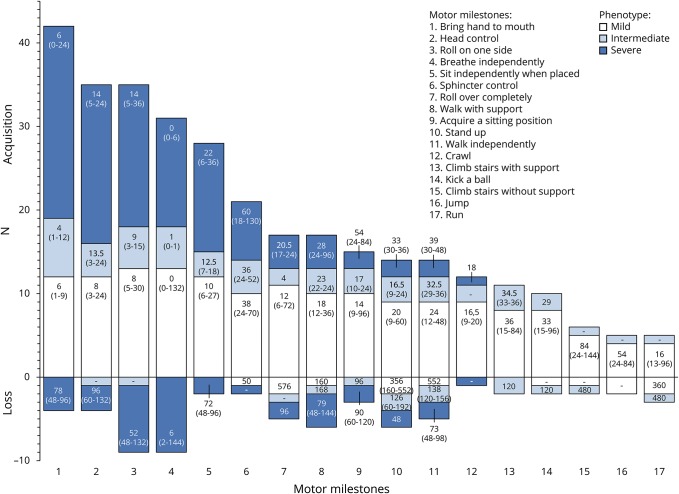
References
-
- Vandersmissen I, Biancalana V, Servais L, et al. An integrated modelling methodology for estimating the prevalence of centronuclear myopathy. Neuromuscul Disord 2018;28:766–777. - PubMed
-
- Hnia K, Vaccari I, Bolino A, Laporte J. Myotubularin phosphoinositide phosphatases: cellular functions and disease pathophysiology. Trends Mol Med 2012;18:317–327. - PubMed
-
- Amoasii L, Hnia K, Laporte J. Myotubularin phosphoinositide phosphatases in human diseases. Curr Top Microbiol Immunol 2012;362:209–233. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical