

Epigenetic therapy of Prader-Willi syndrome
- PMID: 30904443
- PMCID: PMC6527448
- DOI: 10.1016/j.trsl.2019.02.012
Epigenetic therapy of Prader-Willi syndrome
Abstract
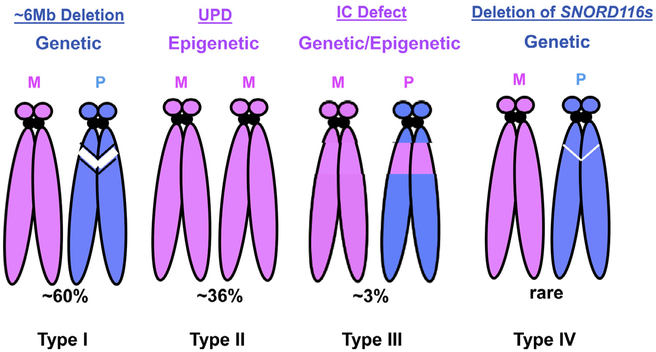
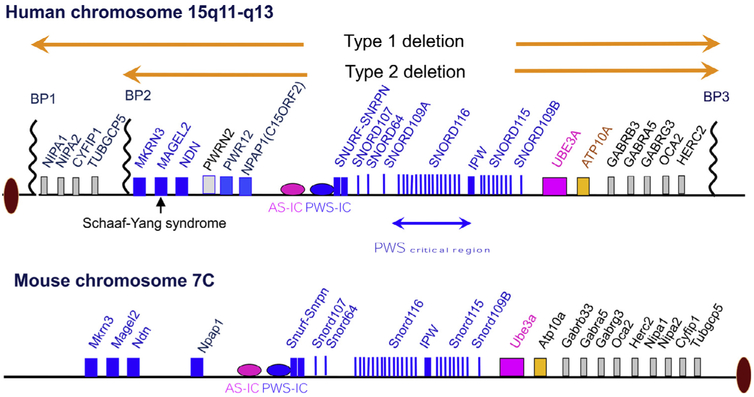
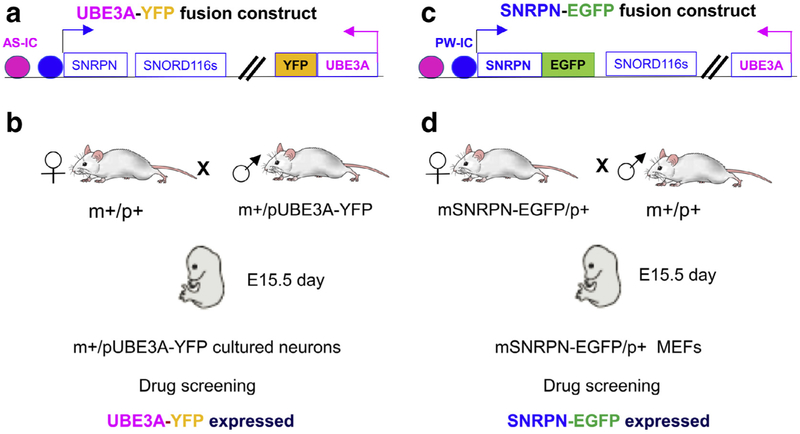
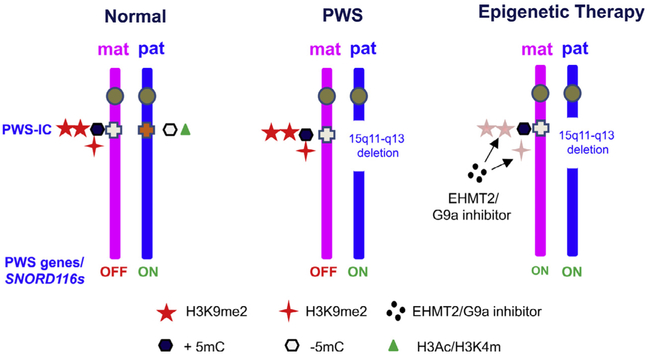
Prader-Willi syndrome (PWS) is a complex and multisystem neurobehavioral disorder. The molecular mechanism of PWS is deficiency of paternally expressed gene gene or genes from the chromosome 15q11-q13. Due to imprinted gene regulation, the same genes in the maternal chromosome 15q11-q13 are structurally intact but transcriptionally repressed by an epigenetic mechanism. The unique molecular defect underlying PWS renders an exciting opportunity to explore epigenetic-based therapy to reactivate the expression of repressed PWS genes from the maternal chromosome. Inactivation of H3K9m3 methyltransferase SETDB1 and zinc finger protein ZNF274 results in reactivation of SNRPN and SNORD116 cluster from the maternal chromosomes in PWS patient iPSCs and iPSC-derived neurons, respectively. High content screening of small molecule libraries using cells derived from transgenic mice carrying the SNRPN-EGFP fusion protein has discovered that inhibitors of EHMT2/G9a, a histone 3 lysine 9 methyltransferase, are capable of reactivating expression of paternally expressed SNRPN and SNORD116 from the maternal chromosome, both in cultured PWS patient-derived fibroblasts and in a PWS mouse model. Treatment with an EMHT2/G9a inhibitor also rescues perinatal lethality and failure to thrive phenotypes in a PWS mouse model. These findings present the first evidence to support a proof-of-principle for epigenetic-based therapy for the PWS in humans.
Copyright © 2019 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Reactivation of maternal SNORD116 cluster via SETDB1 knockdown in Prader-Willi syndrome iPSCs.Hum Mol Genet. 2014 Sep 1;23(17):4674-85. doi: 10.1093/hmg/ddu187. Epub 2014 Apr 23. Hum Mol Genet. 2014. PMID: 24760766 Free PMC article.
-
Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome.Nat Med. 2017 Feb;23(2):213-222. doi: 10.1038/nm.4257. Epub 2016 Dec 26. Nat Med. 2017. PMID: 28024084 Free PMC article.
-
RNAi Knockdown of EHMT2 in Maternal Expression of Prader-Willi Syndrome Genes.Genes (Basel). 2024 Oct 24;15(11):1366. doi: 10.3390/genes15111366. Genes (Basel). 2024. PMID: 39596566 Free PMC article.
-
Prader-Willi Syndrome: Molecular Mechanism and Epigenetic Therapy.Curr Gene Ther. 2020;20(1):36-43. doi: 10.2174/1566523220666200424085336. Curr Gene Ther. 2020. PMID: 32329685 Review.
-
Cognitive deficits in the Snord116 deletion mouse model for Prader-Willi syndrome.Neurobiol Learn Mem. 2019 Nov;165:106874. doi: 10.1016/j.nlm.2018.05.011. Epub 2018 May 23. Neurobiol Learn Mem. 2019. PMID: 29800646 Free PMC article. Review.
Cited by
-
The RDoC approach for translational psychiatry: Could a genetic disorder with psychiatric symptoms help fill the matrix? the example of Prader-Willi syndrome.Transl Psychiatry. 2020 Aug 8;10(1):274. doi: 10.1038/s41398-020-00964-6. Transl Psychiatry. 2020. PMID: 32772048 Free PMC article. Review.
-
Epigenetics meets GPCR: inhibition of histone H3 methyltransferase (G9a) and histamine H3 receptor for Prader-Willi Syndrome.Sci Rep. 2020 Aug 11;10(1):13558. doi: 10.1038/s41598-020-70523-y. Sci Rep. 2020. PMID: 32782417 Free PMC article.
-
The Pivotal Role of Oxytocin's Mechanism of Thermoregulation in Prader-Willi Syndrome, Schaaf-Yang Syndrome, and Autism Spectrum Disorder.Int J Mol Sci. 2024 Feb 8;25(4):2066. doi: 10.3390/ijms25042066. Int J Mol Sci. 2024. PMID: 38396741 Free PMC article. Review.
-
Progress in Brain Magnetic Resonance Imaging of Individuals with Prader-Willi Syndrome.J Clin Med. 2023 Jan 29;12(3):1054. doi: 10.3390/jcm12031054. J Clin Med. 2023. PMID: 36769704 Free PMC article. Review.
-
Molecular and Clinical Opposite Findings in 11p15.5 Associated Imprinting Disorders: Characterization of Basic Mechanisms to Improve Clinical Management.Int J Mol Sci. 2019 Aug 28;20(17):4219. doi: 10.3390/ijms20174219. Int J Mol Sci. 2019. PMID: 31466347 Free PMC article. Review.
References
-
- Ledbetter DH, Riccardi VM, Airhart SD, Strobel RJ, Keenan BS, Crawford JD. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med 1981;304:325–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
