

False signals induced by single-cell imputation
- PMID: 30906525
- PMCID: PMC6415334
- DOI: 10.12688/f1000research.16613.2
False signals induced by single-cell imputation
Abstract
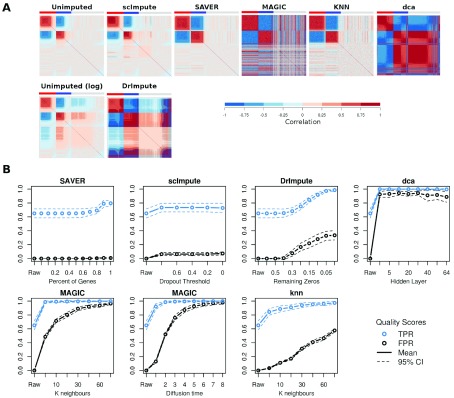
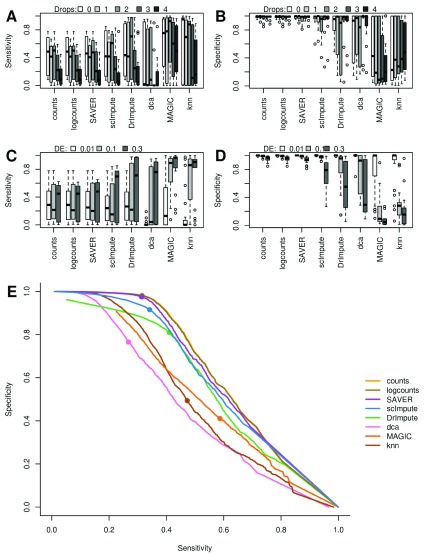
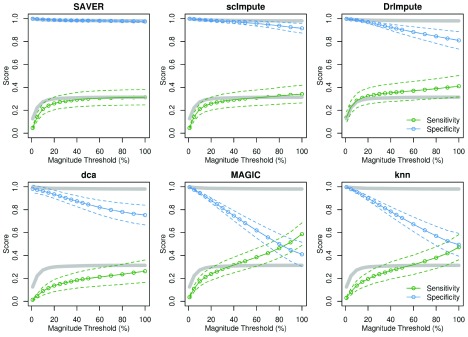
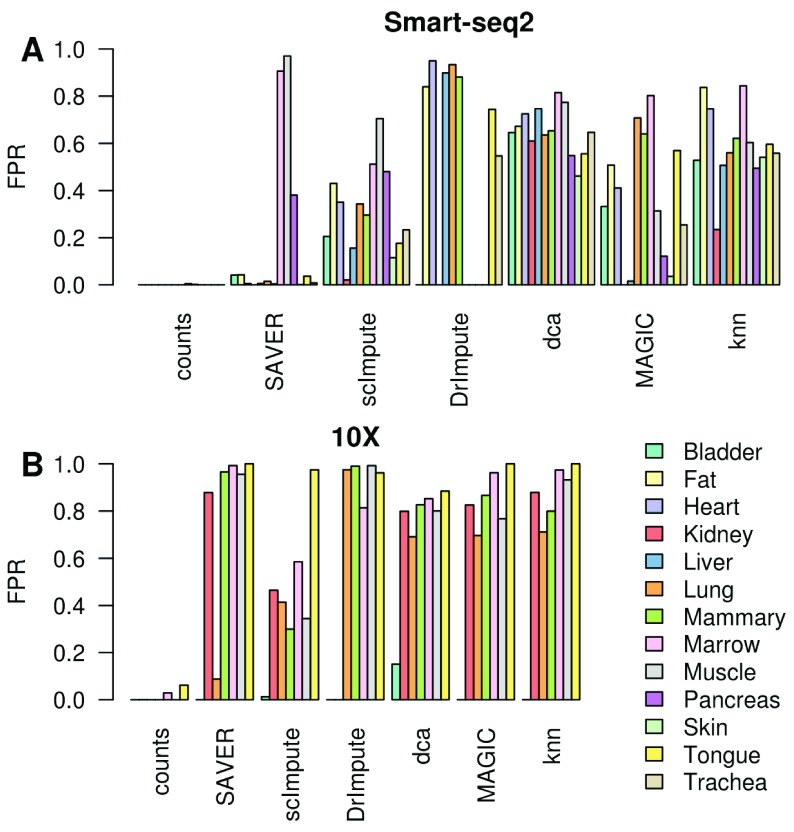
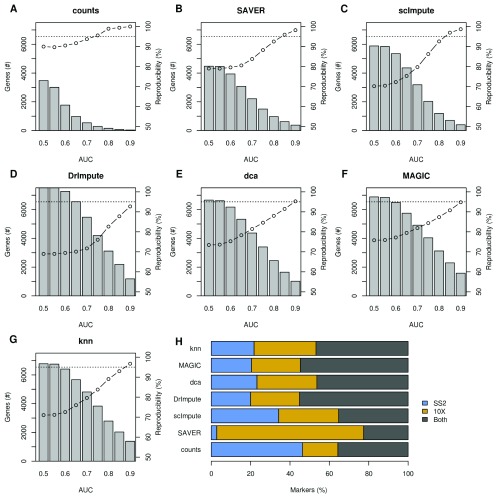
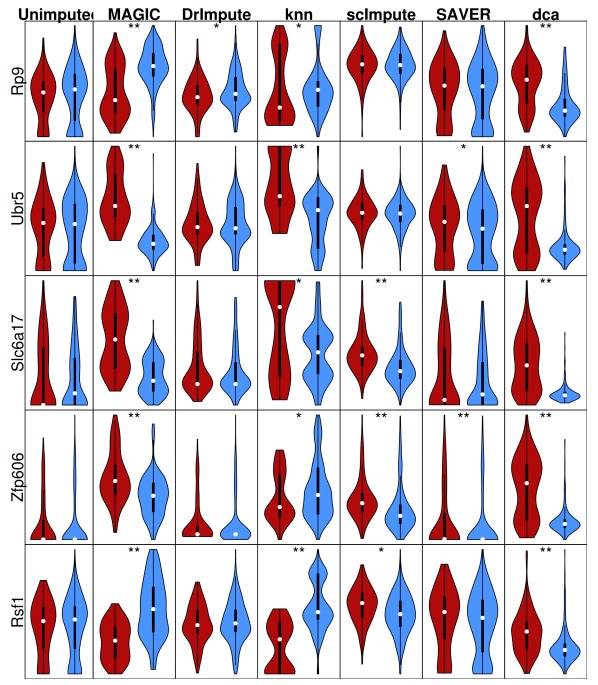
Background: Single-cell RNA-seq is a powerful tool for measuring gene expression at the resolution of individual cells. A challenge in the analysis of this data is the large amount of zero values, representing either missing data or no expression. Several imputation approaches have been proposed to address this issue, but they generally rely on structure inherent to the dataset under consideration they may not provide any additional information, hence, are limited by the information contained therein and the validity of their assumptions. Methods: We evaluated the risk of generating false positive or irreproducible differential expression when imputing data with six different methods. We applied each method to a variety of simulated datasets as well as to permuted real single-cell RNA-seq datasets and consider the number of false positive gene-gene correlations and differentially expressed genes. Using matched 10X and Smart-seq2 data we examined whether cell-type specific markers were reproducible across datasets derived from the same tissue before and after imputation. Results: The extent of false-positives introduced by imputation varied considerably by method. Data smoothing based methods, MAGIC, knn-smooth and dca, generated many false-positives in both real and simulated data. Model-based imputation methods typically generated fewer false-positives but this varied greatly depending on the diversity of cell-types in the sample. All imputation methods decreased the reproducibility of cell-type specific markers, although this could be mitigated by selecting markers with large effect size and significance. Conclusions: Imputation of single-cell RNA-seq data introduces circularity that can generate false-positive results. Thus, statistical tests applied to imputed data should be treated with care. Additional filtering by effect size can reduce but not fully eliminate these effects. Of the methods we considered, SAVER was the least likely to generate false or irreproducible results, thus should be favoured over alternatives if imputation is necessary.
Keywords: Gene expression; Imputation; RNA-seq; Reproducibility; Type 1 errors; single-cell.
Conflict of interest statement
No competing interests were disclosed.
Figures
References
-
- Benjamini Y, Hochberg Y: Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Roy Stat Soc B Met. 1995;57(1):289–300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
-
- Consortium, The Tabula Muris: Single-cell RNA-seq data from Smart-seq2 sequencing of FACS sorted cells. figshare.Fileset.2017a. 10.6084/m9.figshare.5715040.v1 - DOI
-
- Consortium, The Tabula Muris: Single-cell RNA-seq data from Smart-seq2 sequencing of FACS sorted cells. figshare.Fileset.2017b. 10.6084/m9.figshare.5715040.v1 - DOI
Publication types
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources