

Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer's disease
- PMID: 30906861
- PMCID: PMC6426469
- DOI: 10.1126/sciadv.aav0316
Newly developed reversible MAO-B inhibitor circumvents the shortcomings of irreversible inhibitors in Alzheimer's disease
Abstract
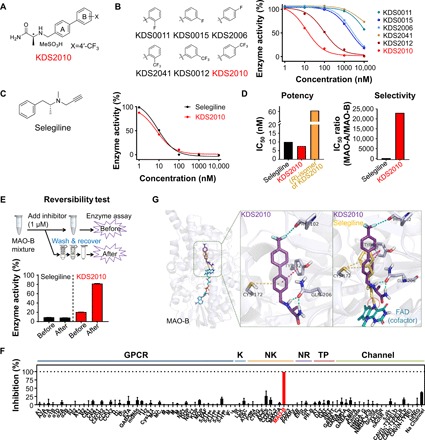
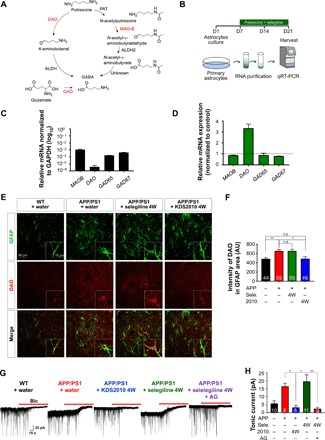
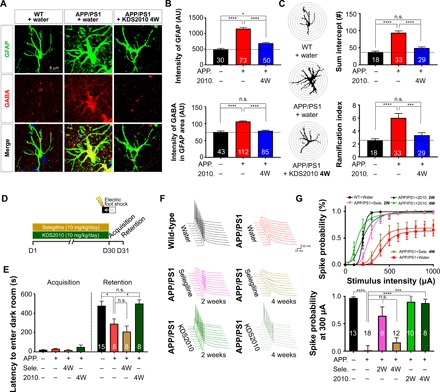
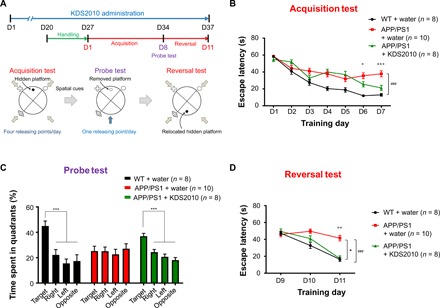
Monoamine oxidase-B (MAO-B) has recently emerged as a potential therapeutic target for Alzheimer's disease (AD) because of its association with aberrant γ-aminobutyric acid (GABA) production in reactive astrocytes. Although short-term treatment with irreversible MAO-B inhibitors, such as selegiline, improves cognitive deficits in AD patients, long-term treatments have shown disappointing results. We show that prolonged treatment with selegiline fails to reduce aberrant astrocytic GABA levels and rescue memory impairment in APP/PS1 mice, an animal model of AD, because of increased activity in compensatory genes for a GABA-synthesizing enzyme, diamine oxidase (DAO). We have developed a potent, highly selective, and reversible MAO-B inhibitor, KDS2010 (IC50 = 7.6 nM; 12,500-fold selectivity over MAO-A), which overcomes the disadvantages of the irreversible MAO-B inhibitor. Long-term treatment with KDS2010 does not induce compensatory mechanisms, thereby significantly attenuating increased astrocytic GABA levels and astrogliosis, enhancing synaptic transmission, and rescuing learning and memory impairments in APP/PS1 mice.
Figures

References
-
- Alzheimer's Association , 2012 Alzheimer's disease facts and figures. Alzheimers Dement. 8, 131–168 (2012). - PubMed
-
- Alzheimer's Association , 2015 Alzheimer's disease facts and figures. Alzheimers Dement. 11, 332–384 (2015). - PubMed
-
- Querfurth H. W., LaFerla F. M., Alzheimer’s Disease. N. Engl. J. Med. 362, 329–344 (2010). - PubMed
-
- Heneka M. T., Carson M. J., Khoury J. E., Landreth G. E., Brosseron F., Feinstein D. L., Jacobs A. H., Wyss-Coray T., Vitorica J., Ransohoff R. M., Herrup K., Frautschy S. A., Finsen B., Brown G. C., Verkhratsky A., Yamanaka K., Koistinaho J., Latz E., Halle A., Petzold G. C., Town T., Morgan D., Shinohara M. L., Perry V. H., Holmes C., Bazan N. G., Brooks D. J., Hunot S., Joseph B., Deigendesch N., Garaschuk O., Boddeke E., Dinarello C. A., Breitner J. C., Cole G. M., Golenbock D. T., Kummer M. P., Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 14, 388–405 (2015). - PMC - PubMed
-
- Hardy J., Allsop D., Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388 (1991). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
