

Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models
- PMID: 30907729
- PMCID: PMC6450673
- DOI: 10.7554/eLife.45457
Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models
Abstract
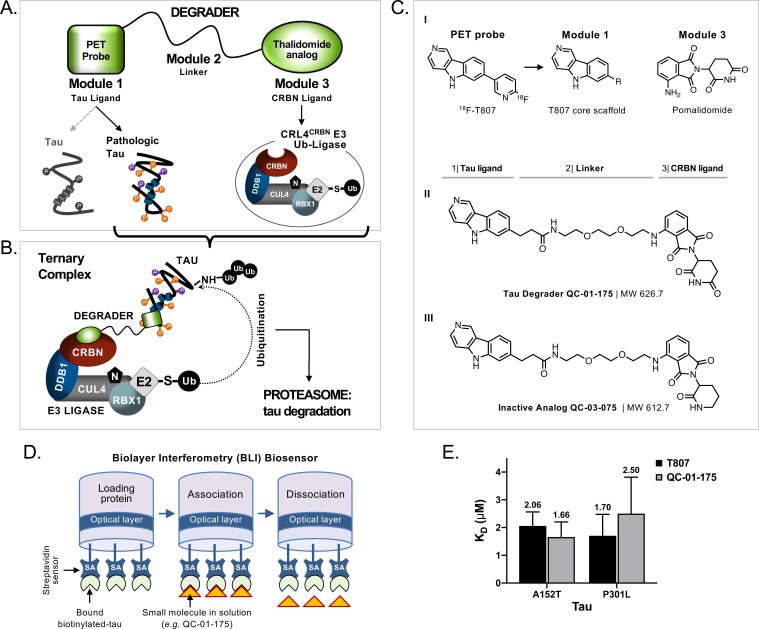
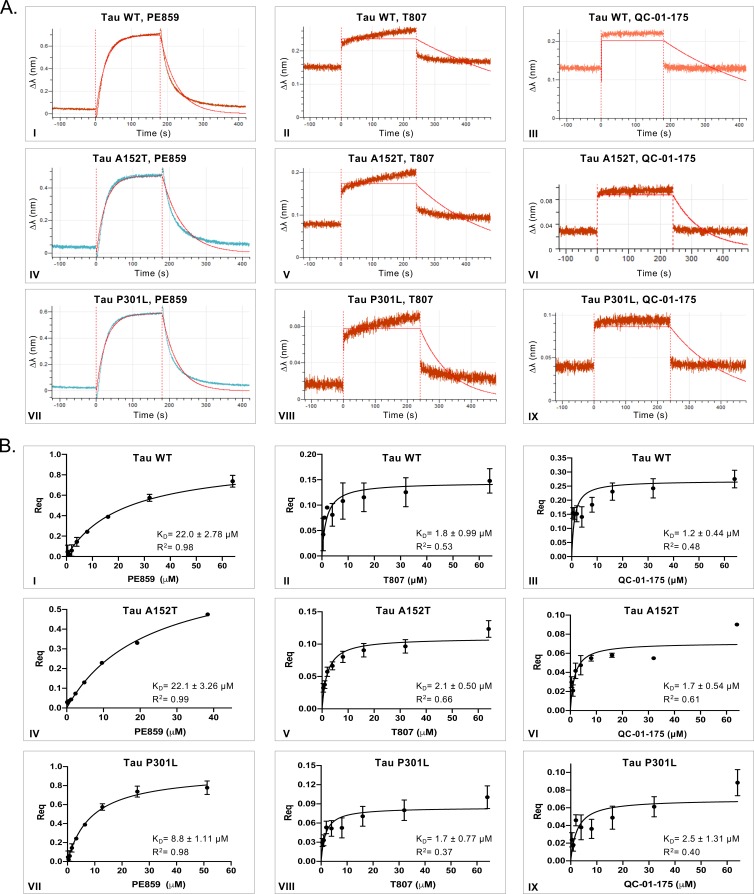
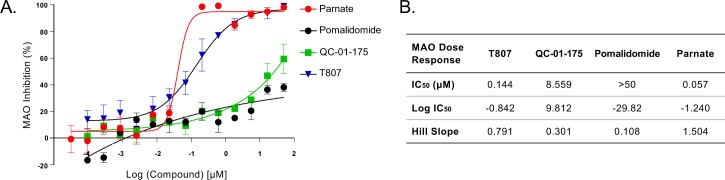
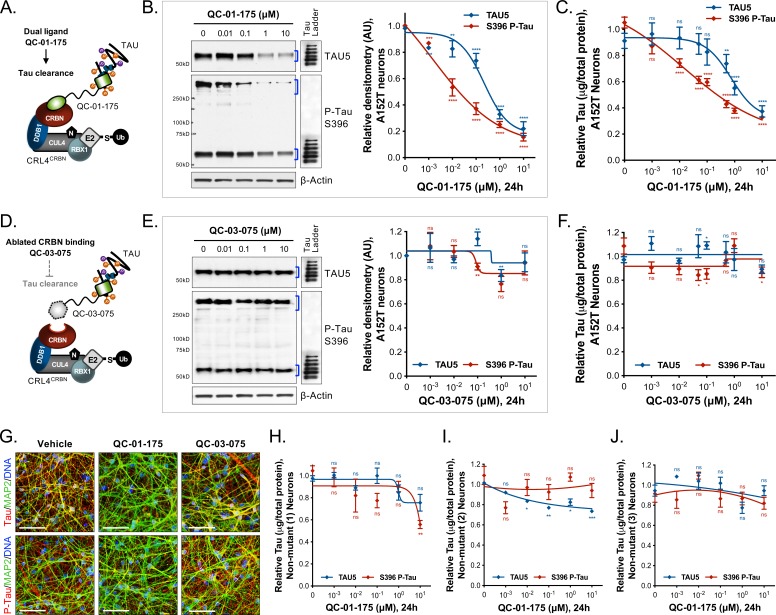
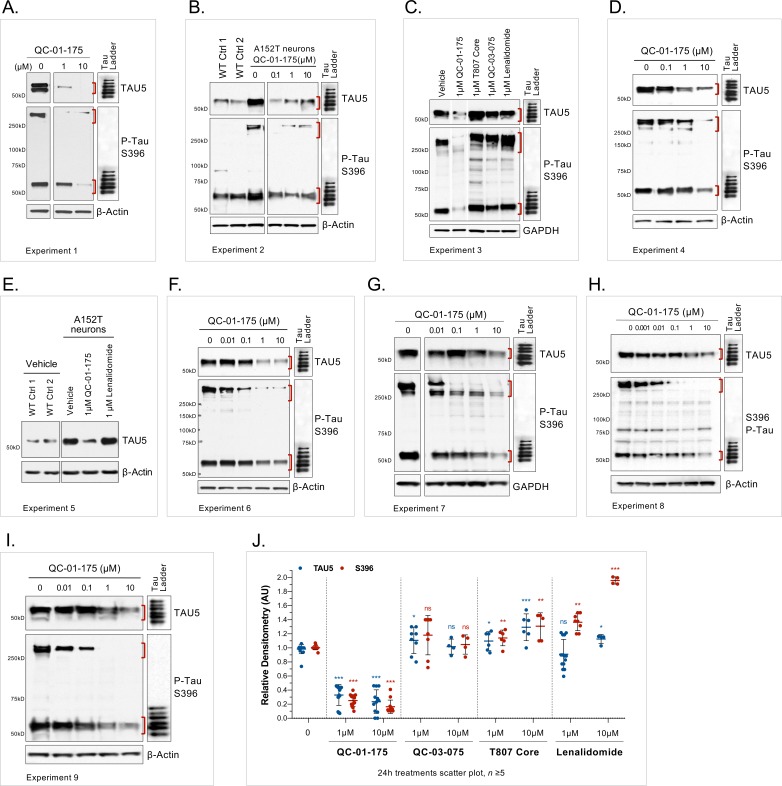
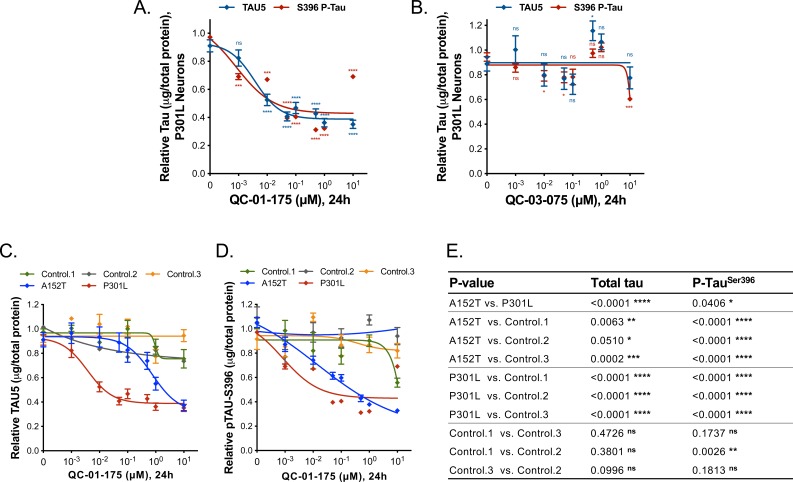
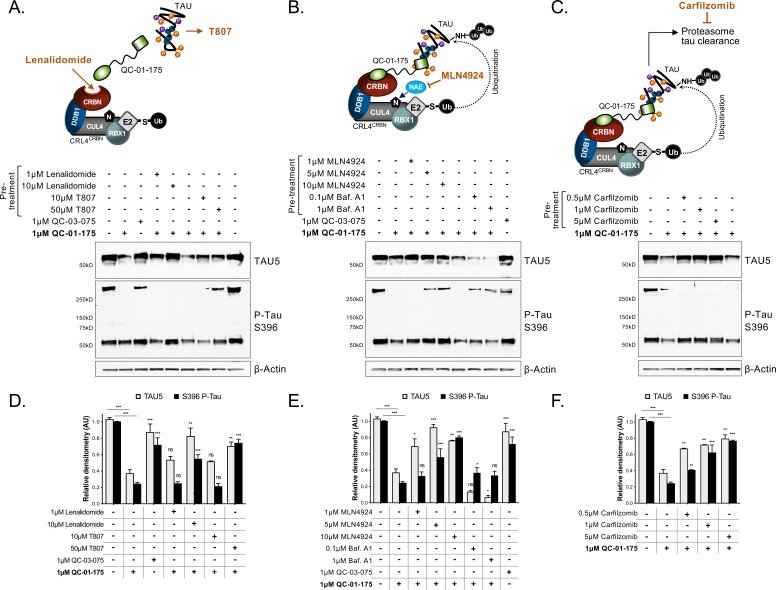
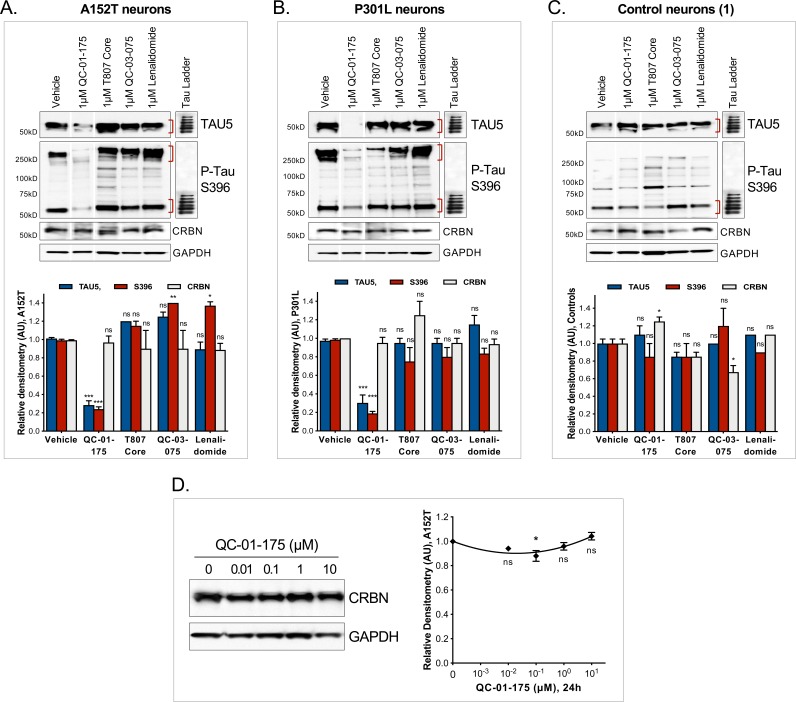
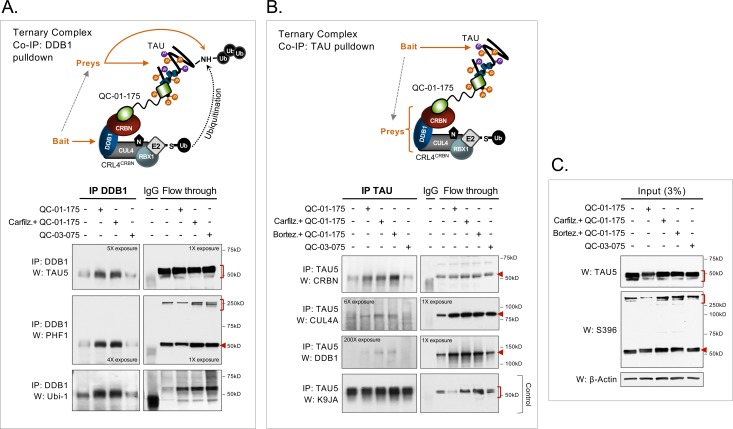
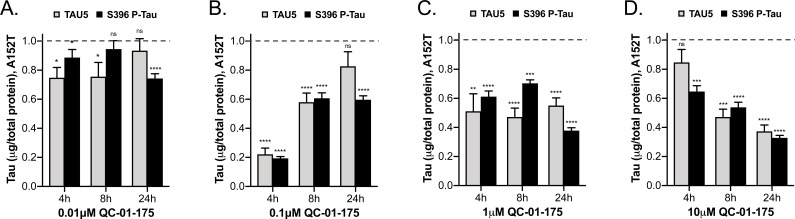
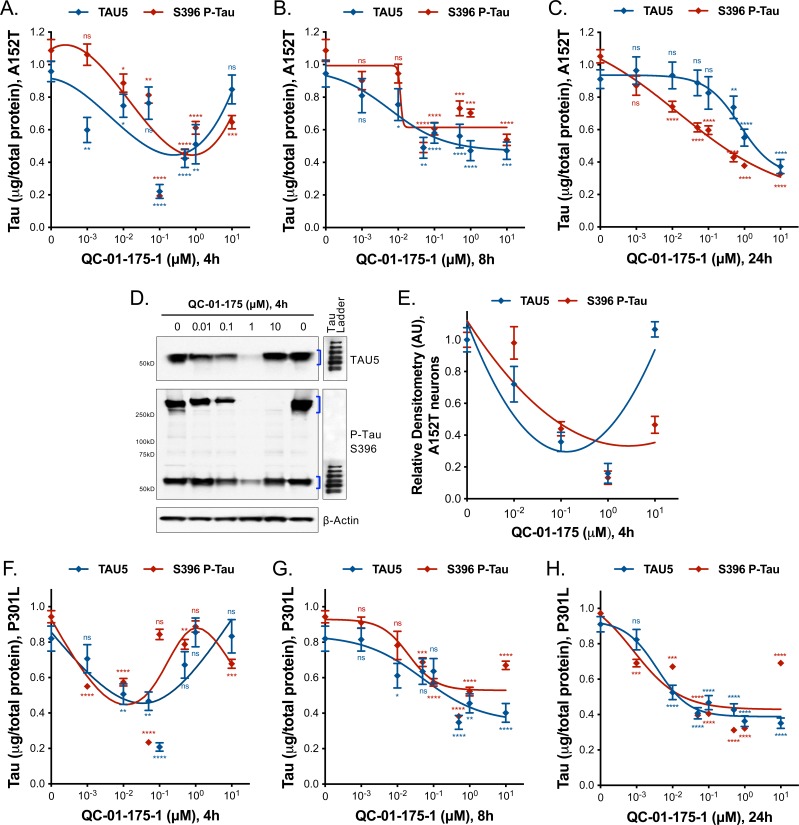
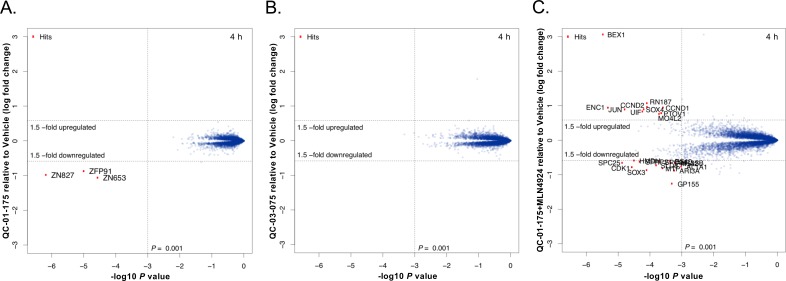
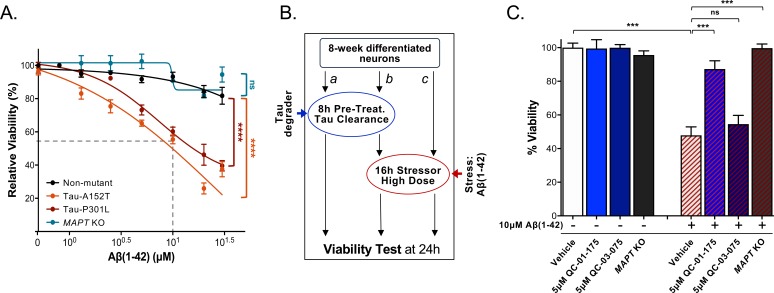
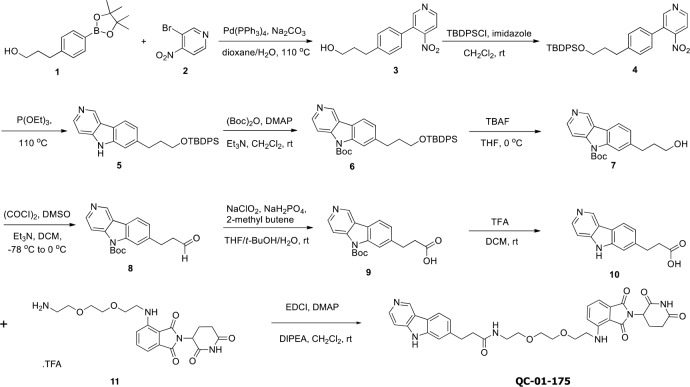
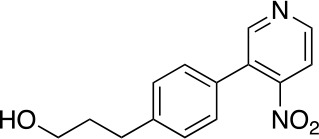
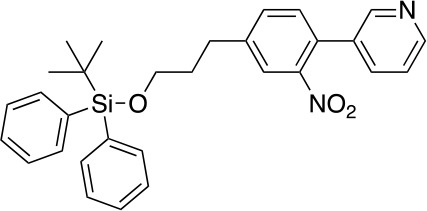
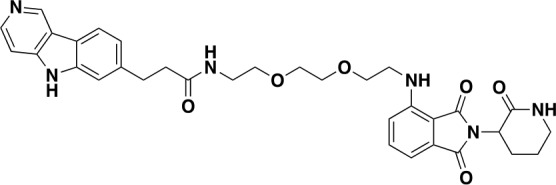
Tauopathies are neurodegenerative diseases characterized by aberrant forms of tau protein accumulation leading to neuronal death in focal brain areas. Positron emission tomography (PET) tracers that bind to pathological tau are used in diagnosis, but there are no current therapies to eliminate these tau species. We employed targeted protein degradation technology to convert a tau PET-probe into a functional degrader of pathogenic tau. The hetero-bifunctional molecule QC-01-175 was designed to engage both tau and Cereblon (CRBN), a substrate-receptor for the E3-ubiquitin ligase CRL4CRBN, to trigger tau ubiquitination and proteasomal degradation. QC-01-175 effected clearance of tau in frontotemporal dementia (FTD) patient-derived neuronal cell models, with minimal effect on tau from neurons of healthy controls, indicating specificity for disease-relevant forms. QC-01-175 also rescued stress vulnerability in FTD neurons, phenocopying CRISPR-mediated MAPT-knockout. This work demonstrates that aberrant tau in FTD patient-derived neurons is amenable to targeted degradation, representing an important advance for therapeutics.
Keywords: biochemistry; chemical biology; degrader; frontotemporal dementia; human; human neuronal cell models; neuroscience; tau.
© 2019, Silva et al.
Conflict of interest statement
MS, FF, QC is a co-inventor on a patent covering the molecules disclosed in this publication (WO/2019/014429), KD, GN, DP, TZ, HH, DL, TM No competing interests declared, BD is a consultant for Merck, Lilly, Biogen, and Piramal; and receives royalties from Oxford University Press, Cambridge University Press, and Elsevier, EF is a SAB member and equity holder in C4 Therapeutics and a consultant to Novartis, AbbVie, and Pfizer. The Fischer lab receives or has received research funding from Novartis, Astellas and Deerfield, NG is a founder, science advisory board member (SAB) and equity holder in Gatekeeper, Syros, Petra, C4, B2S and Soltego. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Her2llc, Deerfield and Sanofi. Co-inventor on a patent covering the molecules disclosed in this publication (WO/2019/014429), SH is a member of the SAB and equity holder in Rodin Therapeutics, Psy Therapeutics, and Frequency Therapeutics. His laboratory has received funding from the Tau Consortium, F-Prime Biomedical Research Initiative, AstraZeneca, JW Pharmaceuticals and speaking fees from AstraZeneca, Amgen, Merck. Co-inventor on a patent covering the molecules disclosed in this publication (WO/2019/014429)
Figures

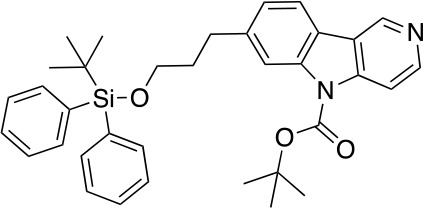
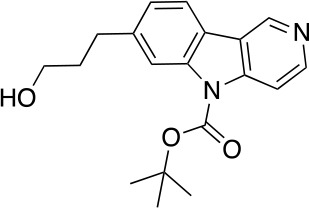
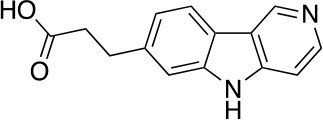
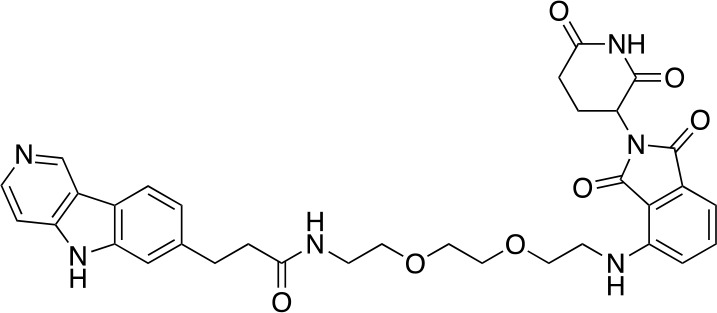
References
-
- Almeida S, Zhang Z, Coppola G, Mao W, Futai K, Karydas A, Geschwind MD, Tartaglia MC, Gao F, Gianni D, Sena-Esteves M, Geschwind DH, Miller BL, Farese RV, Gao FB. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Reports. 2012;2:789–798. doi: 10.1016/j.celrep.2012.09.007. - DOI - PMC - PubMed
-
- Chamberlain PP, Lopez-Girona A, Miller K, Carmel G, Pagarigan B, Chie-Leon B, Rychak E, Corral LG, Ren YJ, Wang M, Riley M, Delker SL, Ito T, Ando H, Mori T, Hirano Y, Handa H, Hakoshima T, Daniel TO, Cathers BE. Structure of the human Cereblon-DDB1-lenalidomide complex reveals basis for responsiveness to thalidomide analogs. Nature Structural & Molecular Biology. 2014;21:803–809. doi: 10.1038/nsmb.2874. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
