

Adenosine kinase attenuates cardiomyocyte microtubule stabilization and protects against pressure overload-induced hypertrophy and LV dysfunction
- PMID: 30910669
- PMCID: PMC6555768
- DOI: 10.1016/j.yjmcc.2019.03.015
Adenosine kinase attenuates cardiomyocyte microtubule stabilization and protects against pressure overload-induced hypertrophy and LV dysfunction
Abstract
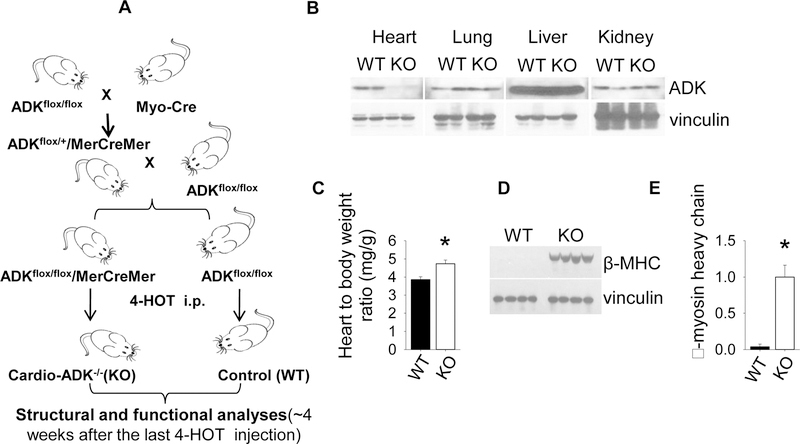
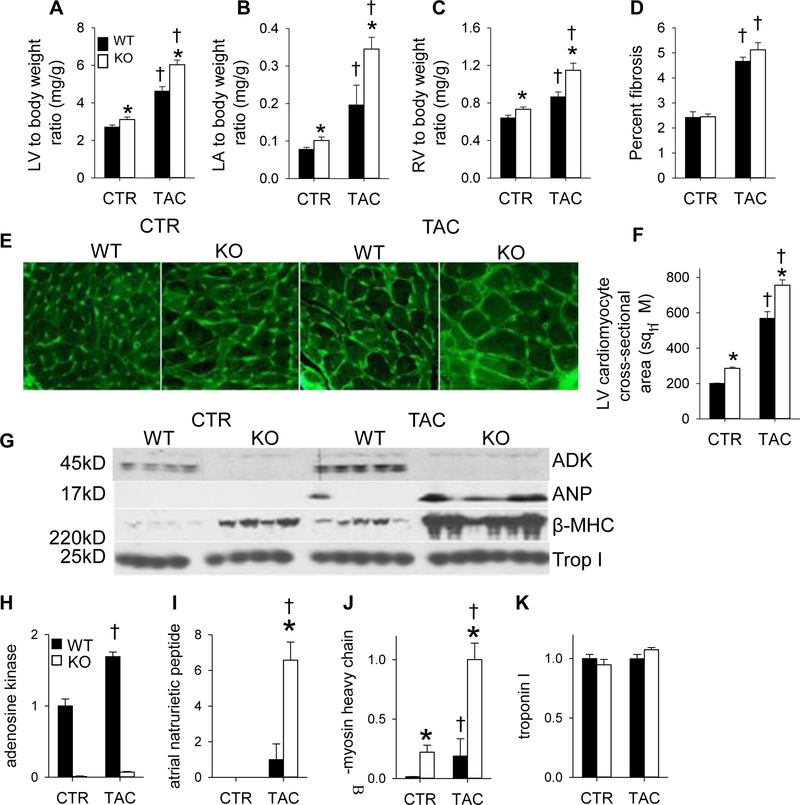
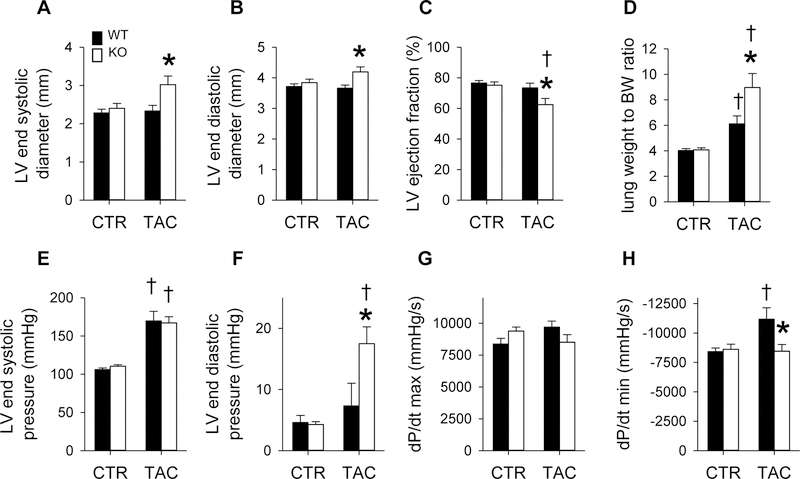
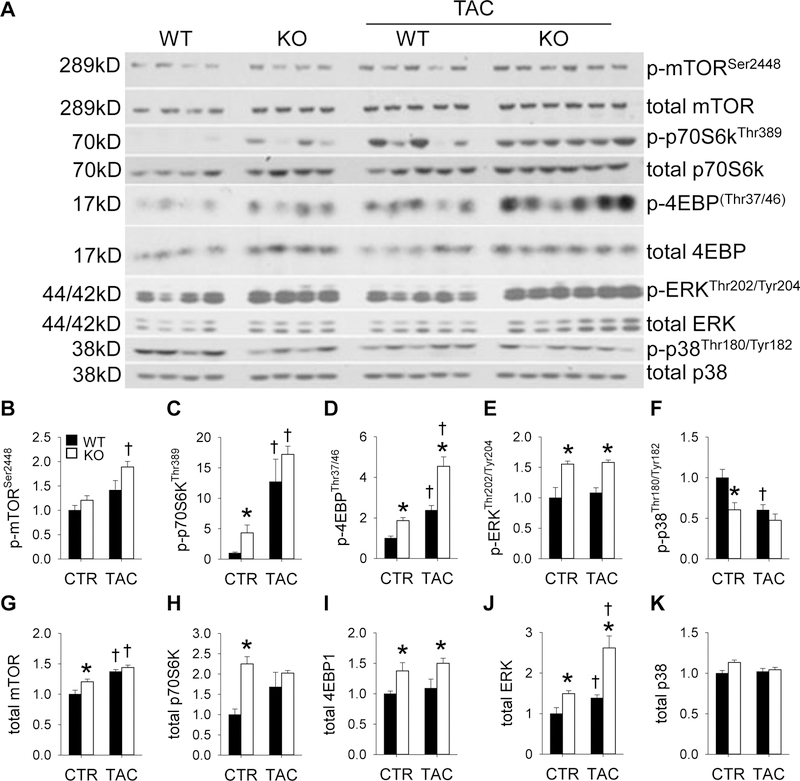
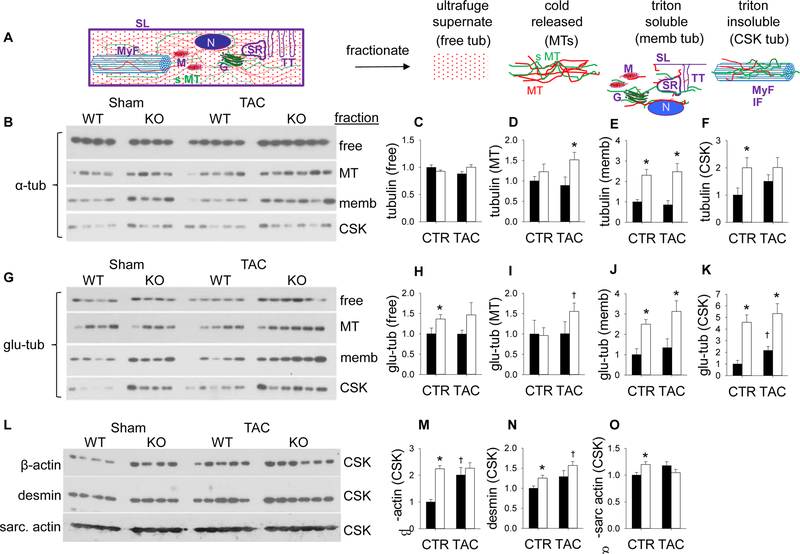
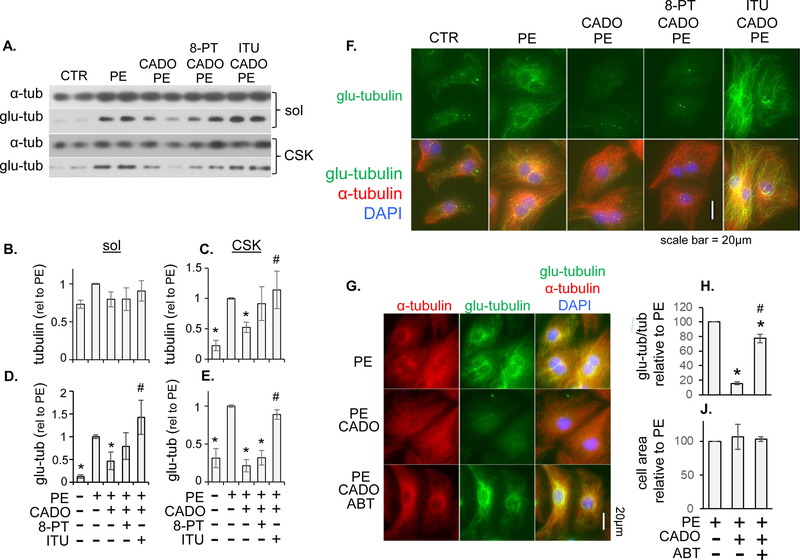
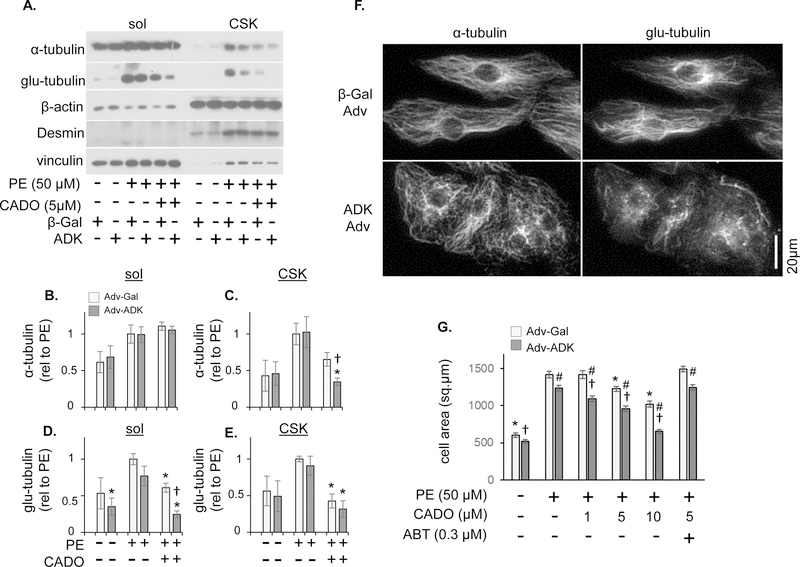
Adenosine exerts numerous protective actions in the heart, including attenuation of cardiac hypertrophy. Adenosine kinase (ADK) converts adenosine to adenosine monophosphate (AMP) and is the major route of myocardial adenosine metabolism, however, the impact of ADK activity on cardiac structure and function is unknown. To examine the role of ADK in cardiac homeostasis and adaptation to stress, conditional cardiomyocyte specific ADK knockout mice (cADK-/-) were produced using the MerCreMer-lox-P system. Within 4 weeks of ADK disruption, cADK-/- mice developed spontaneous hypertrophy and increased β-Myosin Heavy Chain expression without observable LV dysfunction. In response to 6 weeks moderate left ventricular pressure overload (transverse aortic constriction;TAC), wild type mice (WT) exhibited ~60% increase in ventricular ADK expression and developed LV hypertrophy with preserved LV function. In contrast, cADK-/- mice exhibited significantly greater LV hypertrophy and cardiac stress marker expression (atrial natrurietic peptide and β-Myosin Heavy Chain), LV dilation, reduced LV ejection fraction and increased pulmonary congestion. ADK disruption did not decrease protein methylation, inhibit AMPK, or worsen fibrosis, but was associated with persistently elevated mTORC1 and p44/42 ERK MAP kinase signaling and a striking increase in microtubule (MT) stabilization/detyrosination. In neonatal cardiomyocytes exposed to hypertrophic stress, 2-chloroadenosine (CADO) or adenosine treatment suppressed MT detyrosination, which was reversed by ADK inhibition with iodotubercidin or ABT-702. Conversely, adenoviral over-expression of ADK augmented CADO destabilization of MTs and potentiated CADO attenuation of cardiomyocyte hypertrophy. Together, these findings indicate a novel adenosine receptor-independent role for ADK-mediated adenosine metabolism in cardiomyocyte microtubule dynamics and protection against maladaptive hypertrophy.
Keywords: Adenosine; Adenosine kinase; Cardiac hypertrophy; Detyrosinated tubulin; Microtubules.
Copyright © 2019. Published by Elsevier Ltd.
Conflict of interest statement
Figures
Similar articles
-
AMPK attenuates microtubule proliferation in cardiac hypertrophy.Am J Physiol Heart Circ Physiol. 2013 Mar 1;304(5):H749-58. doi: 10.1152/ajpheart.00935.2011. Epub 2013 Jan 11. Am J Physiol Heart Circ Physiol. 2013. PMID: 23316058 Free PMC article.
-
Adenosine regulation of microtubule dynamics in cardiac hypertrophy.Am J Physiol Heart Circ Physiol. 2009 Aug;297(2):H523-32. doi: 10.1152/ajpheart.00462.2009. Epub 2009 Jun 12. Am J Physiol Heart Circ Physiol. 2009. PMID: 19525375 Free PMC article.
-
Ecto-5'-nucleotidase deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction.Hypertension. 2008 Jun;51(6):1557-64. doi: 10.1161/HYPERTENSIONAHA.108.110833. Epub 2008 Apr 7. Hypertension. 2008. PMID: 18391093 Free PMC article.
-
The adenosine kinase hypothesis of epileptogenesis.Prog Neurobiol. 2008 Mar;84(3):249-62. doi: 10.1016/j.pneurobio.2007.12.002. Epub 2007 Dec 23. Prog Neurobiol. 2008. PMID: 18249058 Free PMC article. Review.
-
Adenosine dysfunction in epilepsy.Glia. 2012 Aug;60(8):1234-43. doi: 10.1002/glia.22285. Epub 2011 Dec 22. Glia. 2012. PMID: 22700220 Free PMC article. Review.
Cited by
-
The microtubule cytoskeleton in cardiac mechanics and heart failure.Nat Rev Cardiol. 2022 Jun;19(6):364-378. doi: 10.1038/s41569-022-00692-y. Epub 2022 Apr 19. Nat Rev Cardiol. 2022. PMID: 35440741 Free PMC article. Review.
-
Cardiomyocyte intercellular signalling increases oxidative stress and reprograms the global- and phospho-proteome of cardiac fibroblasts.J Extracell Biol. 2023 Nov 30;2(12):e125. doi: 10.1002/jex2.125. eCollection 2023 Dec. J Extracell Biol. 2023. PMID: 38938901 Free PMC article.
-
Need for Speed: The Importance of Physiological Strain Rates in Determining Myocardial Stiffness.Front Physiol. 2021 Jul 30;12:696694. doi: 10.3389/fphys.2021.696694. eCollection 2021. Front Physiol. 2021. PMID: 34393820 Free PMC article.
-
Cardiomyocyte Microtubules: Control of Mechanics, Transport, and Remodeling.Annu Rev Physiol. 2022 Feb 10;84:257-283. doi: 10.1146/annurev-physiol-062421-040656. Epub 2021 Oct 6. Annu Rev Physiol. 2022. PMID: 34614374 Free PMC article. Review.
-
Inhibition of adenosine kinase attenuates myocardial ischaemia/reperfusion injury.J Cell Mol Med. 2021 Mar;25(6):2931-2943. doi: 10.1111/jcmm.16328. Epub 2021 Feb 1. J Cell Mol Med. 2021. PMID: 33523568 Free PMC article.
References
-
- Solenkova NV, Solodushko V, Cohen MV and Downey JM. Endogenous adenosine protects preconditioned heart during early minutes of reperfusion by activating Akt. Am J Physiol Heart Circ Physiol 2006;290:H441–9. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
