

Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH
- PMID: 30911168
- PMCID: PMC6544023
- DOI: 10.1038/s41586-019-1049-y
Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH
Abstract
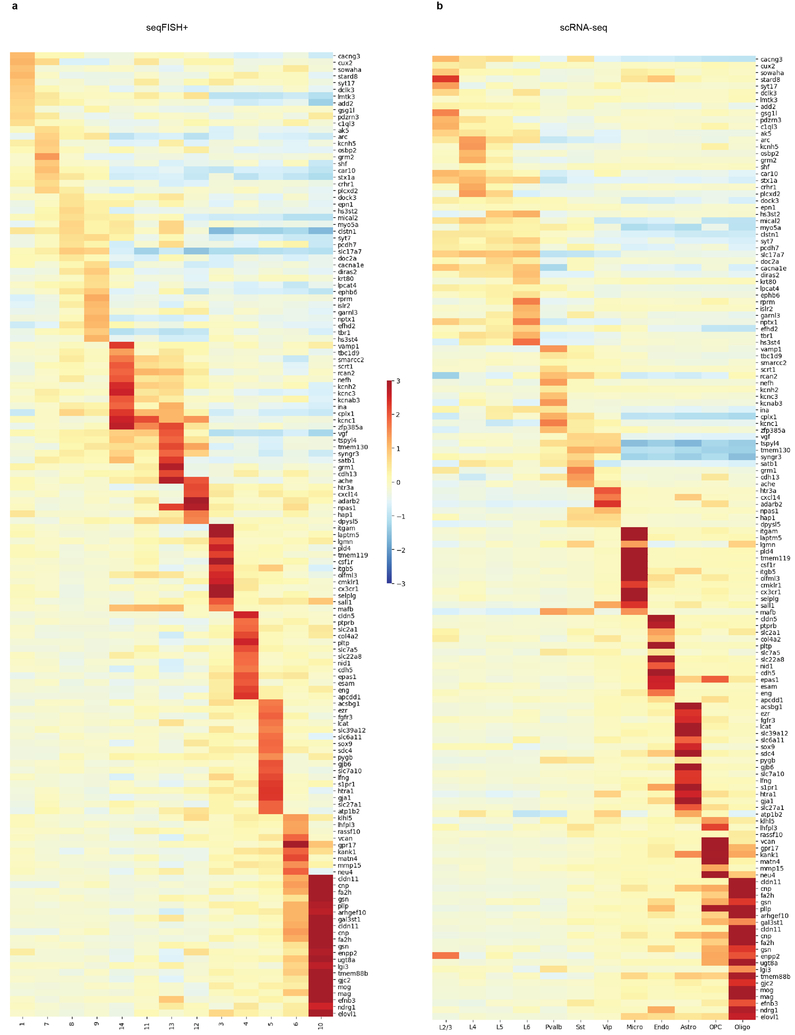
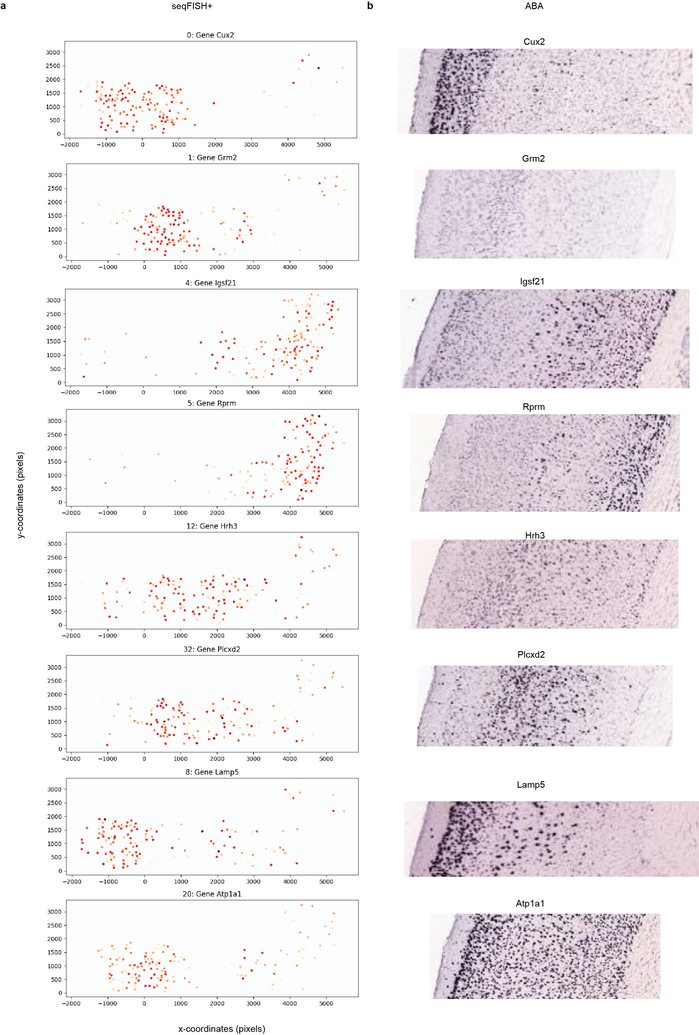
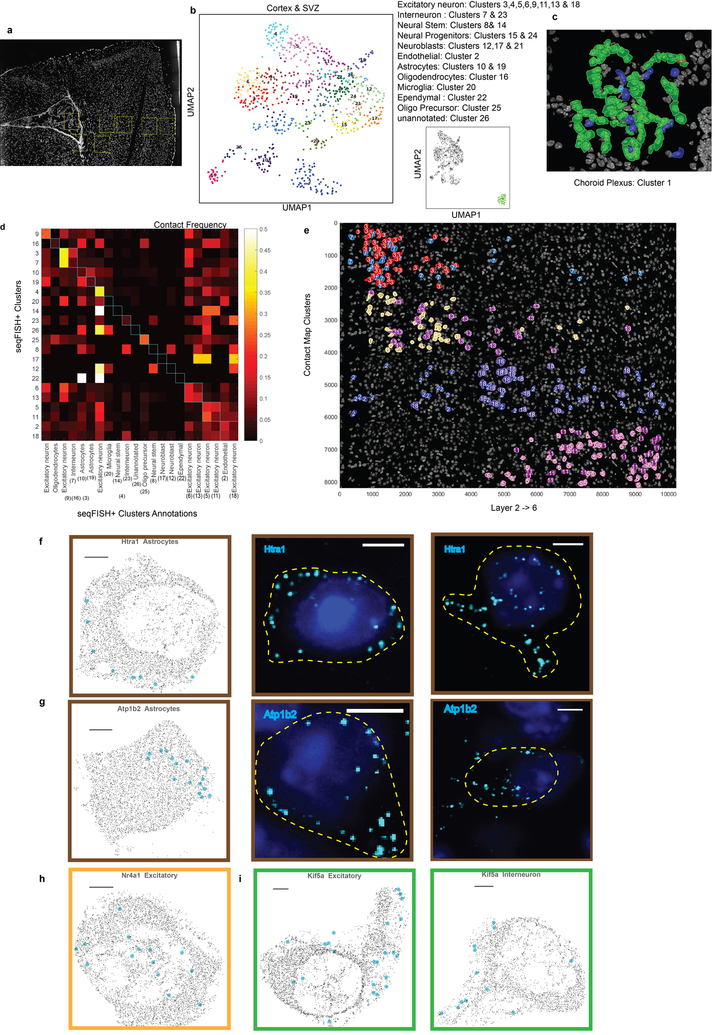
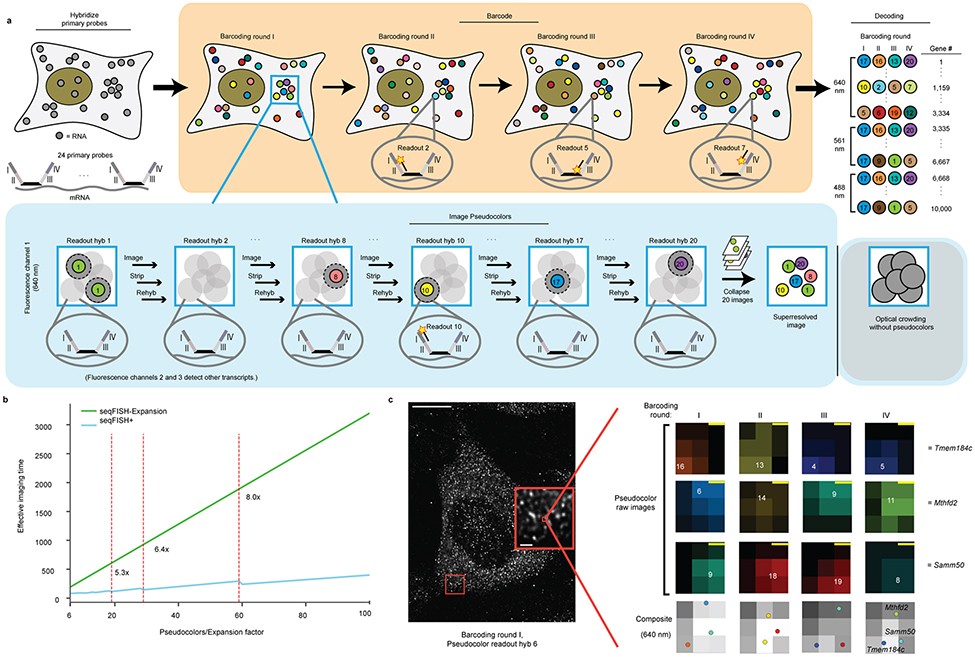
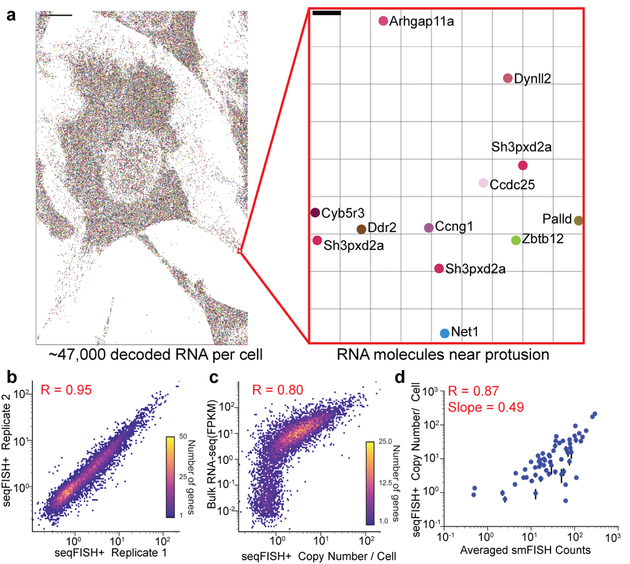
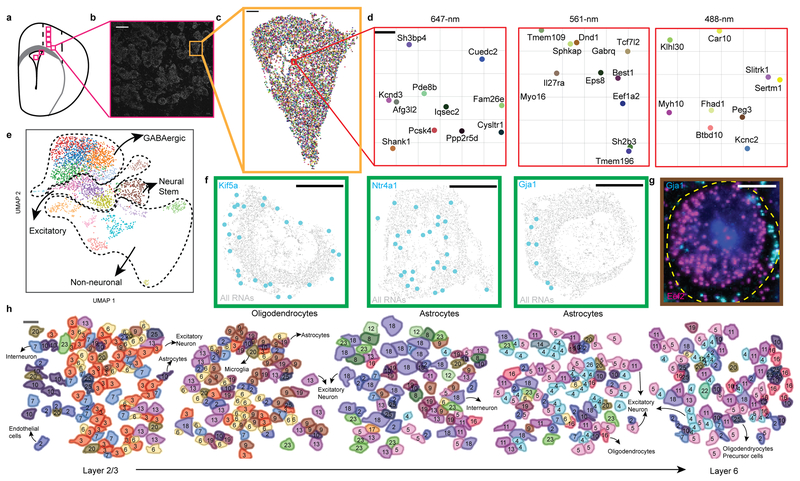
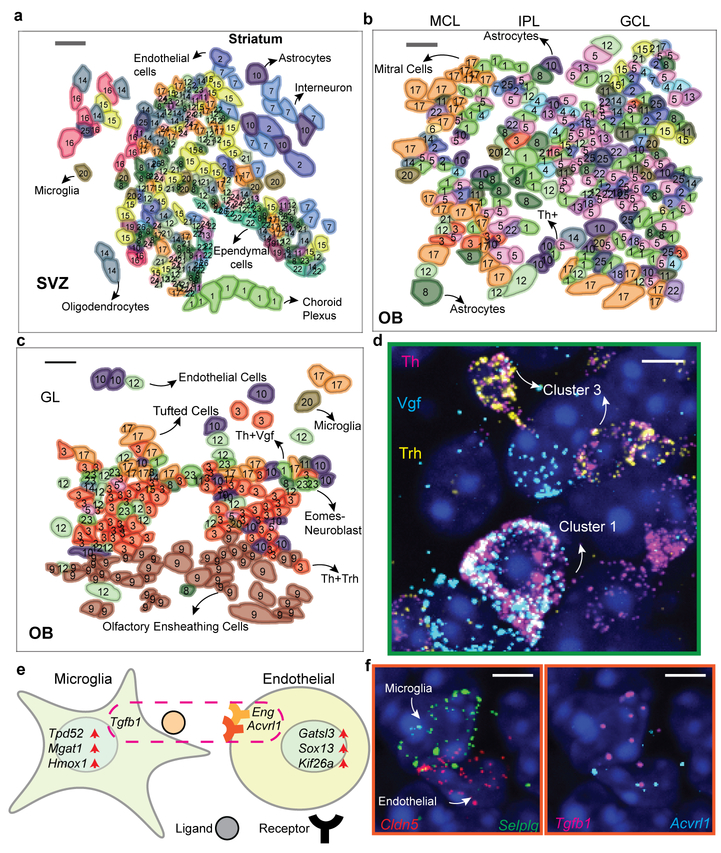
Imaging the transcriptome in situ with high accuracy has been a major challenge in single-cell biology, which is particularly hindered by the limits of optical resolution and the density of transcripts in single cells1-5. Here we demonstrate an evolution of sequential fluorescence in situ hybridization (seqFISH+). We show that seqFISH+ can image mRNAs for 10,000 genes in single cells-with high accuracy and sub-diffraction-limit resolution-in the cortex, subventricular zone and olfactory bulb of mouse brain, using a standard confocal microscope. The transcriptome-level profiling of seqFISH+ allows unbiased identification of cell classes and their spatial organization in tissues. In addition, seqFISH+ reveals subcellular mRNA localization patterns in cells and ligand-receptor pairs across neighbouring cells. This technology demonstrates the ability to generate spatial cell atlases and to perform discovery-driven studies of biological processes in situ.
Conflict of interest statement
Competing Interest:
C.-H.L.E and L.C. filed a patent on the pseudocolor encoding scheme in seqFISH+.
Figures
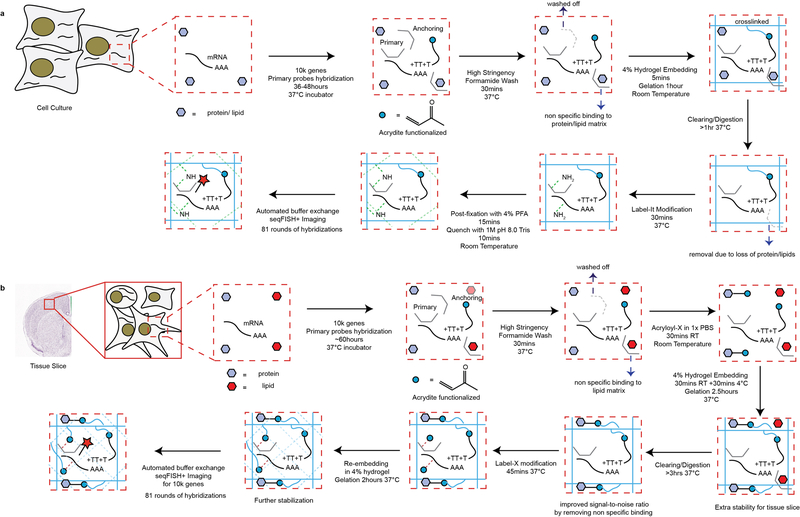
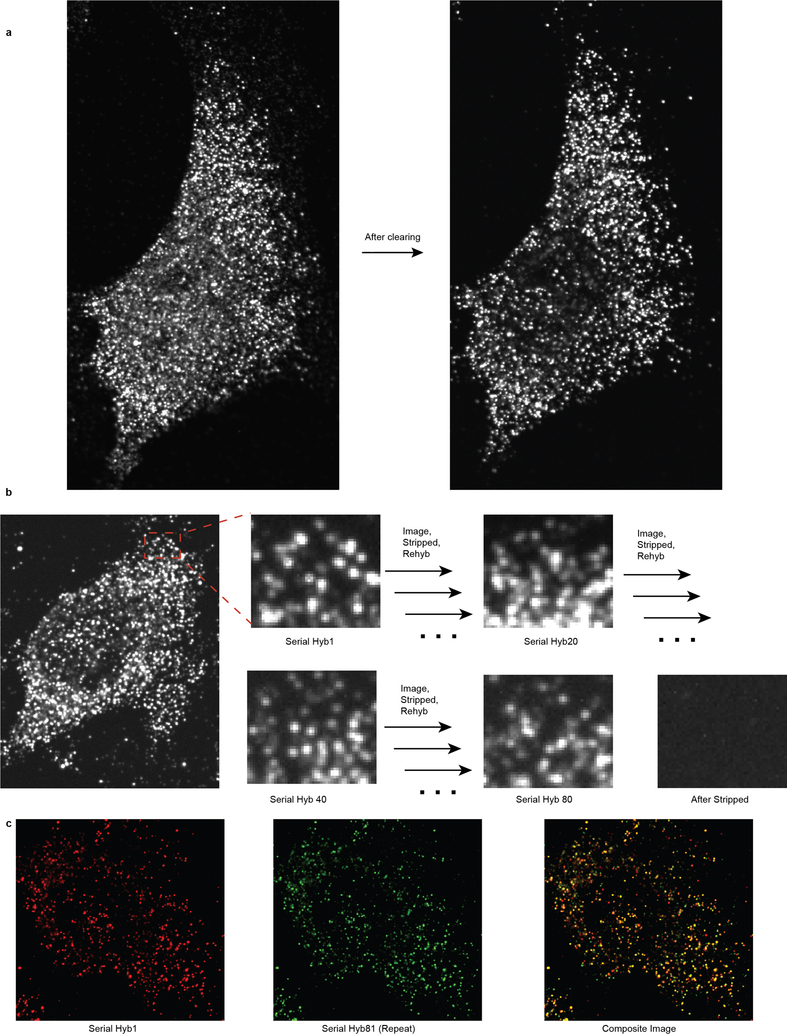
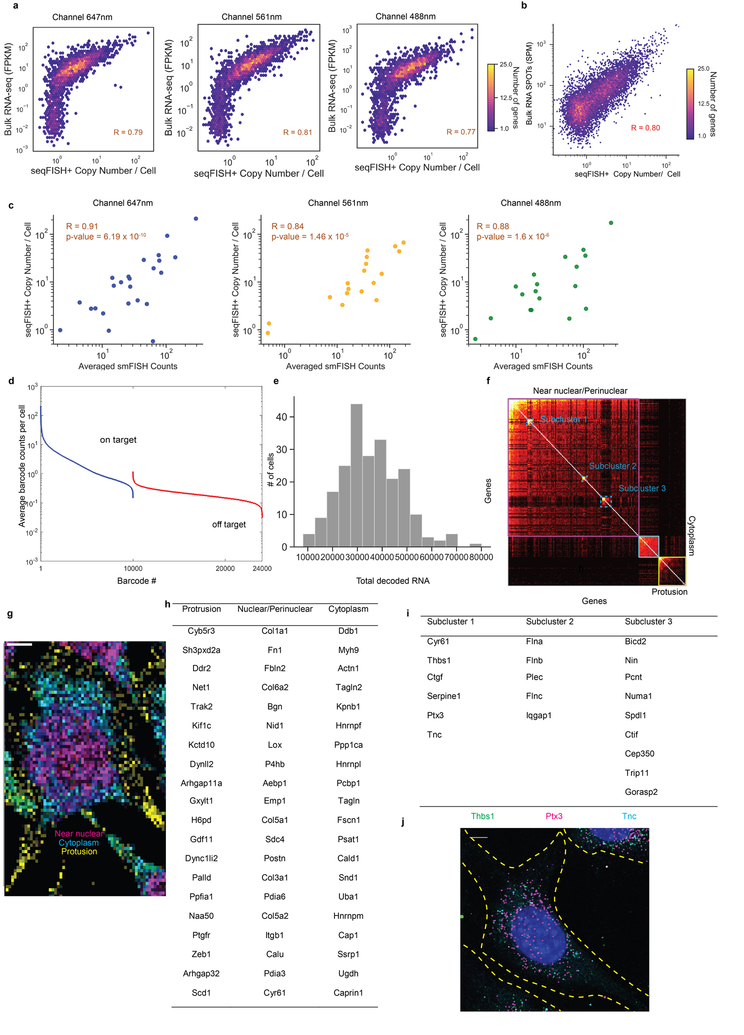
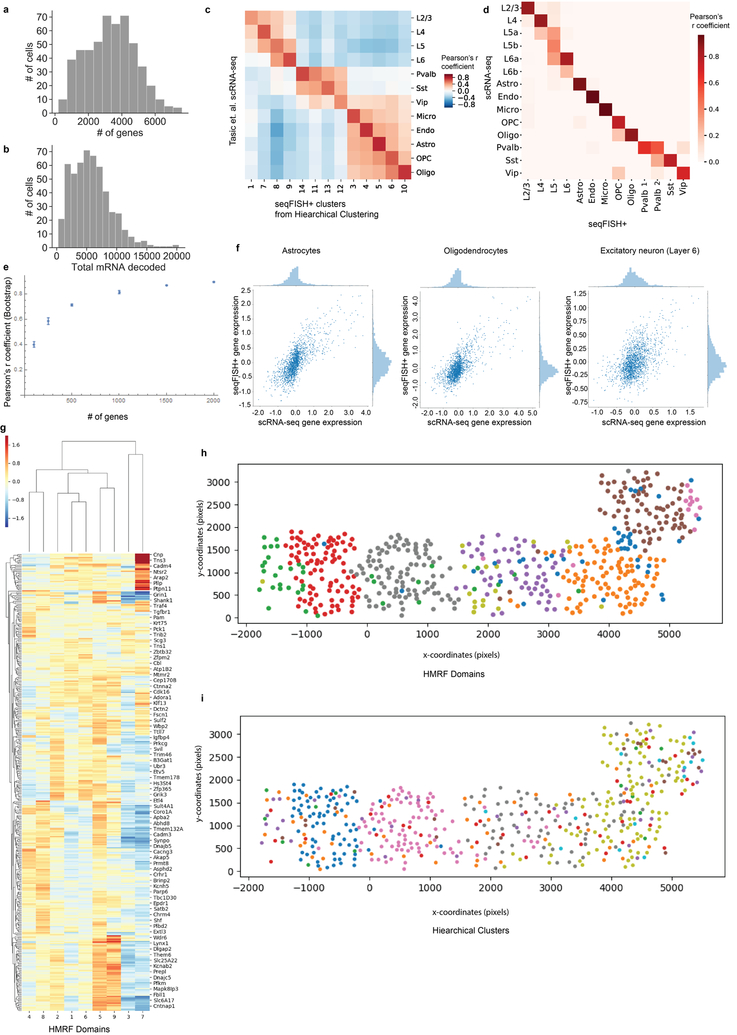



Comment in
-
Spatial transcriptomics coming of age.Nat Rev Genet. 2019 Jun;20(6):317. doi: 10.1038/s41576-019-0129-z. Nat Rev Genet. 2019. PMID: 30980030 No abstract available.
-
A method for transcriptome-wide gene expression quantification in intact tissues.Immunol Cell Biol. 2019 May;97(5):439-441. doi: 10.1111/imcb.12256. Immunol Cell Biol. 2019. PMID: 31131496 No abstract available.
-
Future Directions of Fibrosis in Medicine.Dig Dis. 2022;40(3):299-300. doi: 10.1159/000518039. Epub 2021 Jun 24. Dig Dis. 2022. PMID: 34348286 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
