

Protein damage, ageing and age-related diseases
- PMID: 30914006
- PMCID: PMC6451363
- DOI: 10.1098/rsob.180249
Protein damage, ageing and age-related diseases
Abstract
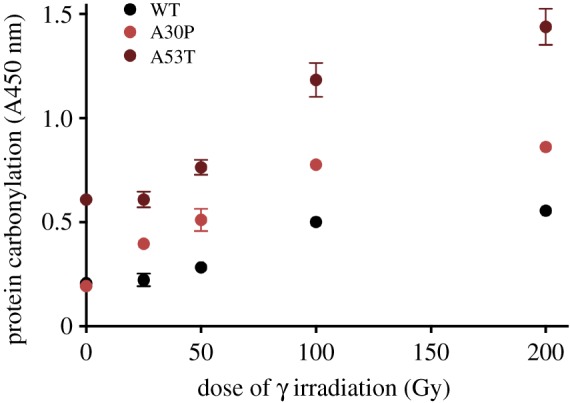
Ageing is considered as a snowballing phenotype of the accumulation of damaged dysfunctional or toxic proteins and silent mutations (polymorphisms) that sensitize relevant proteins to oxidative damage as inborn predispositions to age-related diseases. Ageing is not a disease, but it causes (or shares common cause with) age-related diseases as suggested by similar slopes of age-related increase in the incidence of diseases and death. Studies of robust and more standard species revealed that dysfunctional oxidatively damaged proteins are the root cause of radiation-induced morbidity and mortality. Oxidized proteins accumulate with age and cause reversible ageing-like phenotypes with some irreversible consequences (e.g. mutations). Here, we observe in yeast that aggregation rate of damaged proteins follows the Gompertz law of mortality and review arguments for a causal relationship between oxidative protein damage, ageing and disease. Aerobes evolved proteomes remarkably resistant to oxidative damage, but imperfectly folded proteins become sensitive to oxidation. We show that α-synuclein mutations that predispose to early-onset Parkinson's disease bestow an increased intrinsic sensitivity of α-synuclein to in vitro oxidation. Considering how initially silent protein polymorphism becomes phenotypic while causing age-related diseases and how protein damage leads to genome alterations inspires a vision of predictive diagnostic, prognostic, prevention and treatment of degenerative diseases.
Keywords: age-related diseases; ageing; protein damage.
Conflict of interest statement
We declare we have no competing interests.
Figures







References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases