

Alevin efficiently estimates accurate gene abundances from dscRNA-seq data
- PMID: 30917859
- PMCID: PMC6437997
- DOI: 10.1186/s13059-019-1670-y
Alevin efficiently estimates accurate gene abundances from dscRNA-seq data
Abstract
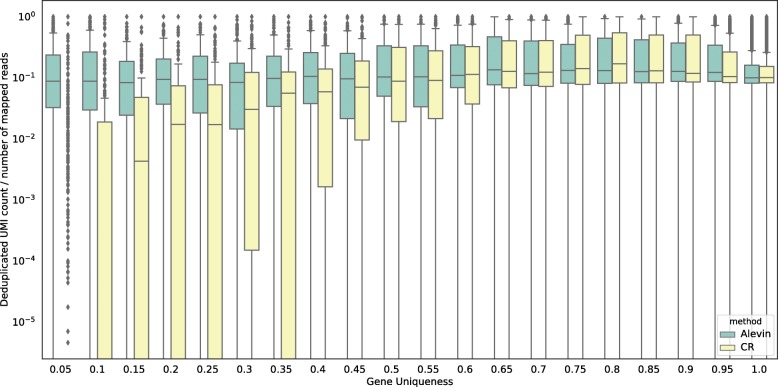
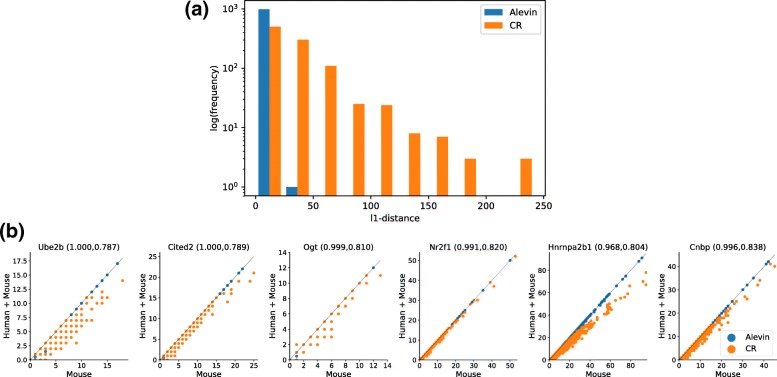
We introduce alevin, a fast end-to-end pipeline to process droplet-based single-cell RNA sequencing data, performing cell barcode detection, read mapping, unique molecular identifier (UMI) deduplication, gene count estimation, and cell barcode whitelisting. Alevin's approach to UMI deduplication considers transcript-level constraints on the molecules from which UMIs may have arisen and accounts for both gene-unique reads and reads that multimap between genes. This addresses the inherent bias in existing tools which discard gene-ambiguous reads and improves the accuracy of gene abundance estimates. Alevin is considerably faster, typically eight times, than existing gene quantification approaches, while also using less memory.
Keywords: Cellular barcode; Quantification; Single-cell RNA-seq; UMI deduplication.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Competing interests
R.P. is a co-founder of Ocean Genomics, Inc. The other authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures






References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
