

Exploring targets of TET2-mediated methylation reprogramming as potential discriminators of prostate cancer progression
- PMID: 30917865
- PMCID: PMC6438015
- DOI: 10.1186/s13148-019-0651-z
Exploring targets of TET2-mediated methylation reprogramming as potential discriminators of prostate cancer progression
Abstract
Background: Global DNA methylation alterations are hallmarks of cancer. The tumor-suppressive TET enzymes, which are involved in DNA demethylation, are decreased in prostate cancer (PCa); in particular, TET2 is specifically targeted by androgen-dependent mechanisms of repression in PCa and may play a central role in carcinogenesis. Thus, the identification of key genes targeted by TET2 dysregulation may provide further insight into cancer biology.
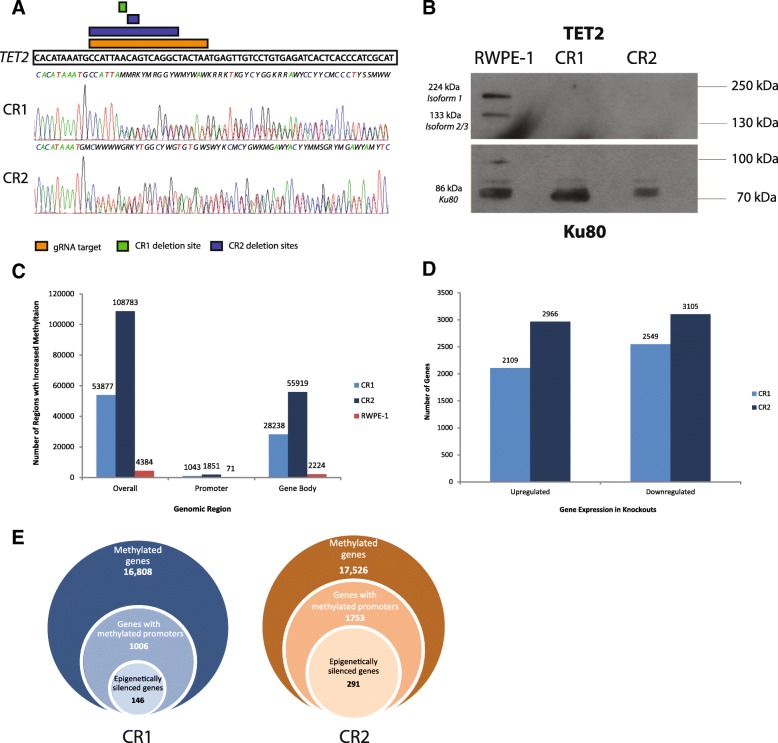
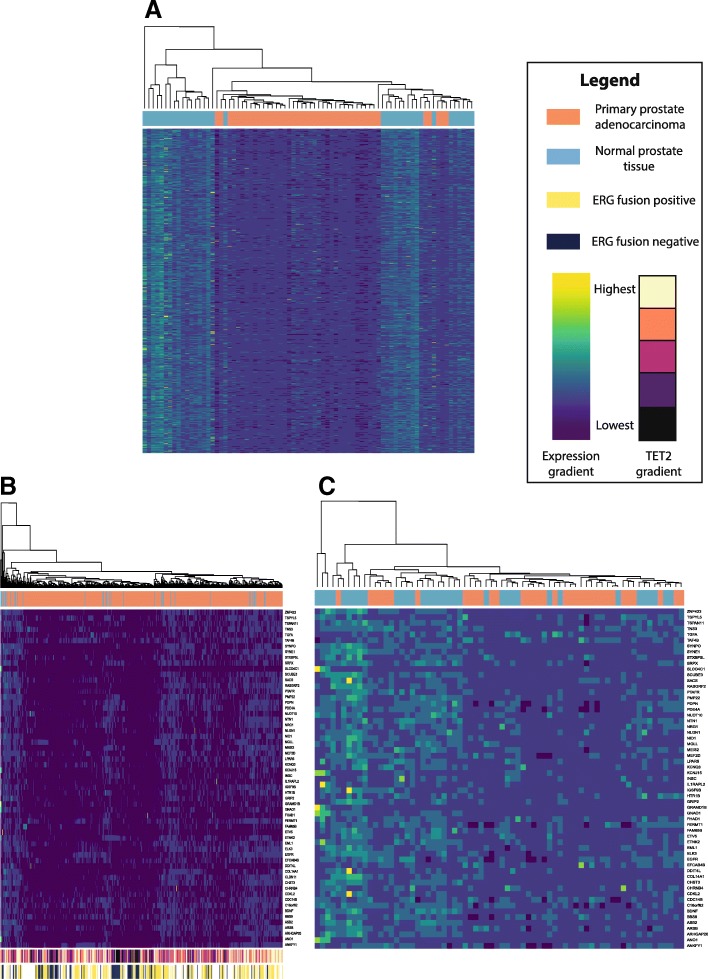
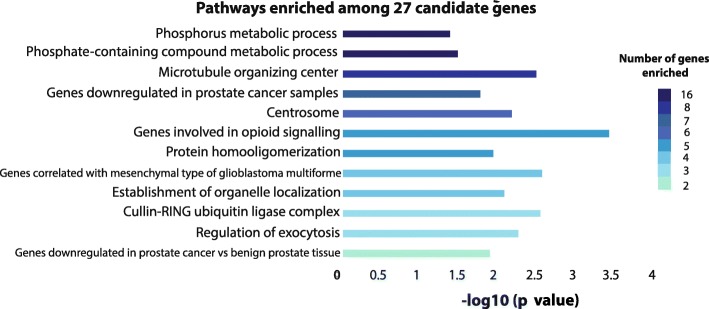
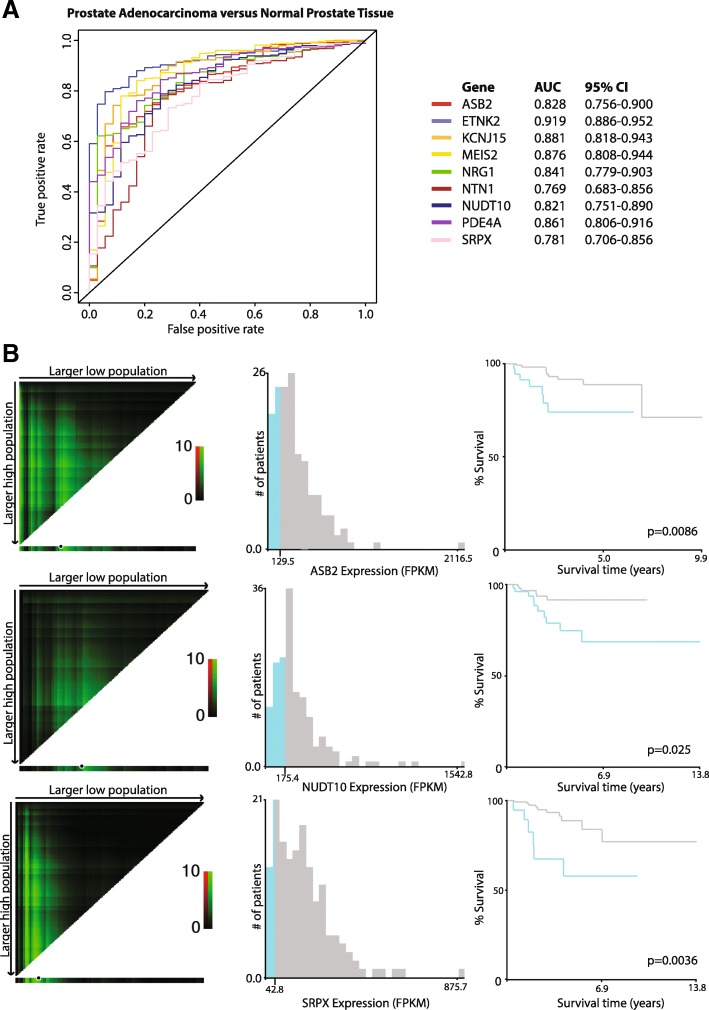
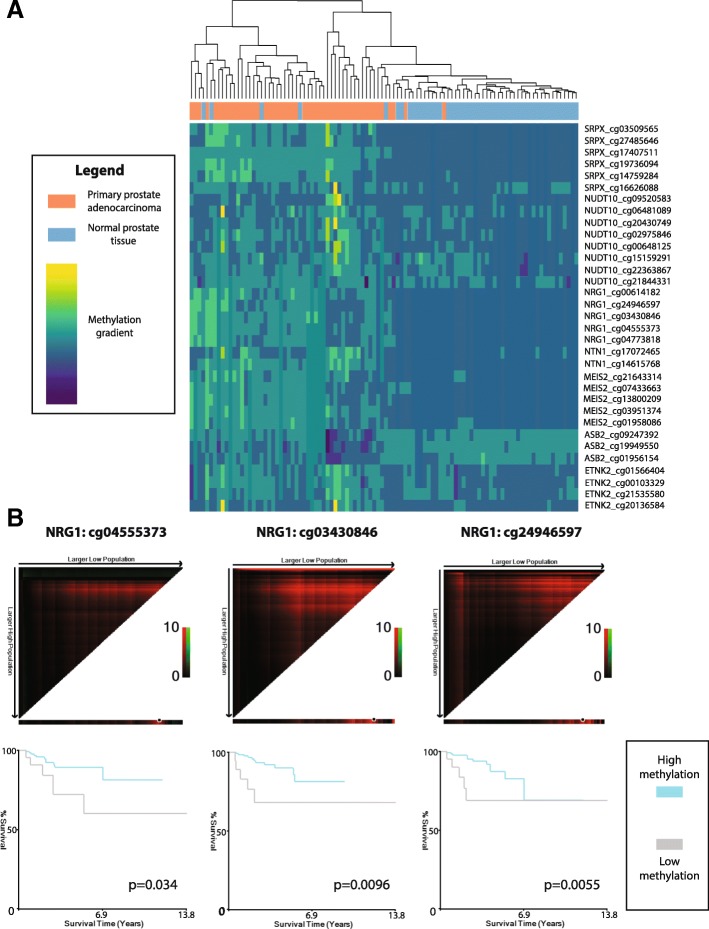
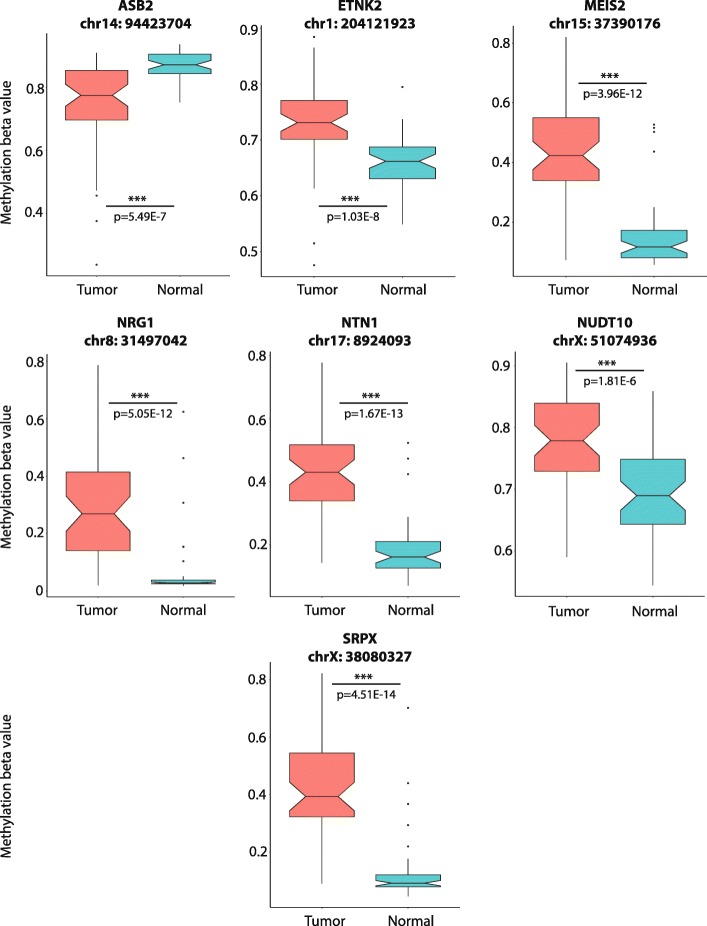
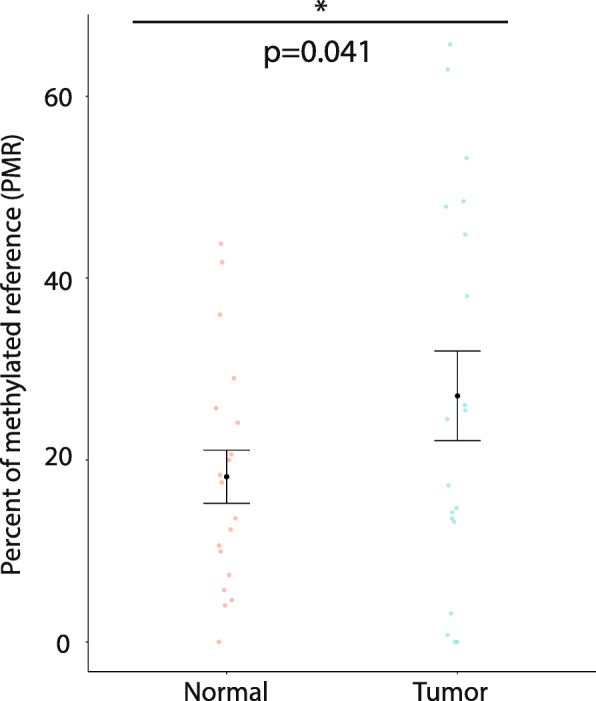
Results: Using a CRISPR/Cas9-derived TET2-knockout prostate cell line, and through whole-transcriptome and whole-methylome sequencing, we identified seven candidate genes-ASB2, ETNK2, MEIS2, NRG1, NTN1, NUDT10, and SRPX-exhibiting reduced expression and increased promoter methylation, a pattern characteristic of tumor suppressors. Decreased expression of these genes significantly discriminates between recurrent and non-recurrent prostate tumors from the Cancer Genome Atlas (TCGA) cohort (n = 423), and ASB2, NUDT10, and SRPX were significantly correlated with lower recurrence-free survival in patients by Kaplan-Meier analysis. ASB2, MEIS2, and SRPX also showed significantly lower expression in high-risk Gleason score 8 tumors as compared to low or intermediate risk tumors, suggesting that these genes may be particularly useful as indicators of PCa progression. Furthermore, methylation array probes in the TCGA dataset, which were proximal to the highly conserved, differentially methylated sites identified in our TET2-knockout cells, were able to significantly distinguish between matched prostate tumor and normal prostate tissues (n = 50 pairs). Except ASB2, all genes exhibited significantly increased methylation at these probes, and methylation status of at least one probe for each of these genes showed association with measures of PCa progression such as recurrence, stage, or Gleason score. Since ASB2 did not have any probes within the TET2-knockout differentially methylated region, we validated ASB2 methylation in an independent series of matched tumor-normal samples (n = 19) by methylation-specific qPCR, which revealed concordant and significant increases in promoter methylation within the TET2-knockout site.
Conclusions: Our study identifies seven genes governed by TET2 loss in PCa which exhibit an association between their methylation and expression status and measures of PCa progression. As differential methylation profiles and TET2 expression are associated with advanced PCa, further investigation of these specialized TET2 targets may provide important insights into patterns of carcinogenic gene dysregulation.
Keywords: Differential methylation profiling; Integrative analysis; Prostate cancer; TET2 knockout; Tumor progression.
Conflict of interest statement
Ethics approval and consent to participate
Matched human prostate tumor and normal tissue was obtained as per ethical approvals established by the Research Ethics Board of Mount Sinai Hospital, Toronto, Ontario (REB reference #140071-E).
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
