

MS1 ion current-based quantitative proteomics: A promising solution for reliable analysis of large biological cohorts
- PMID: 30920002
- PMCID: PMC6849792
- DOI: 10.1002/mas.21595
MS1 ion current-based quantitative proteomics: A promising solution for reliable analysis of large biological cohorts
Abstract
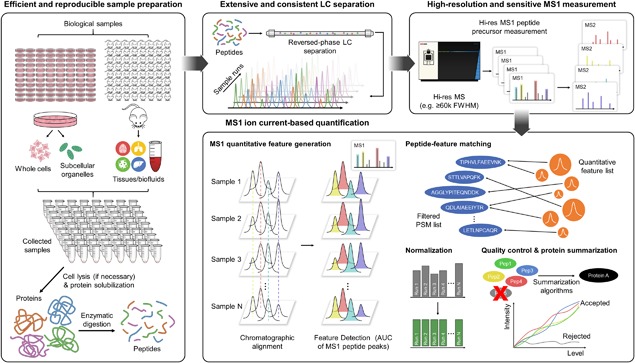
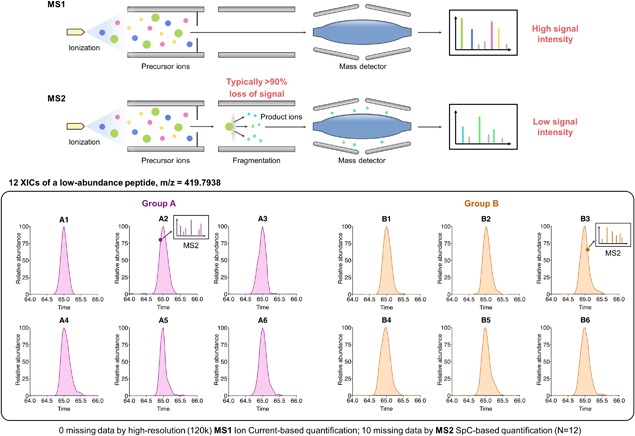
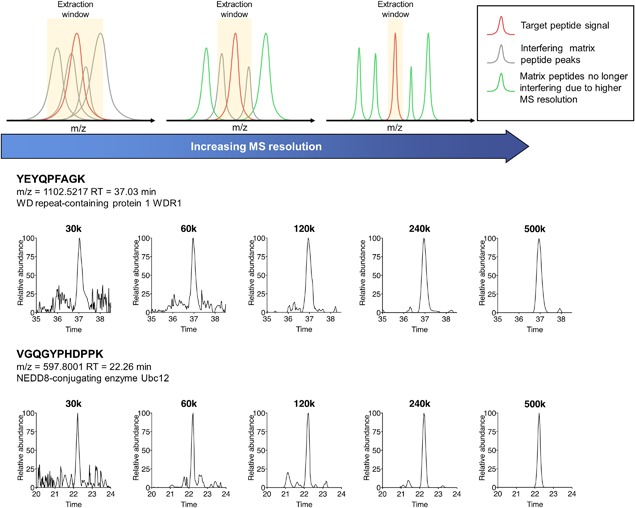
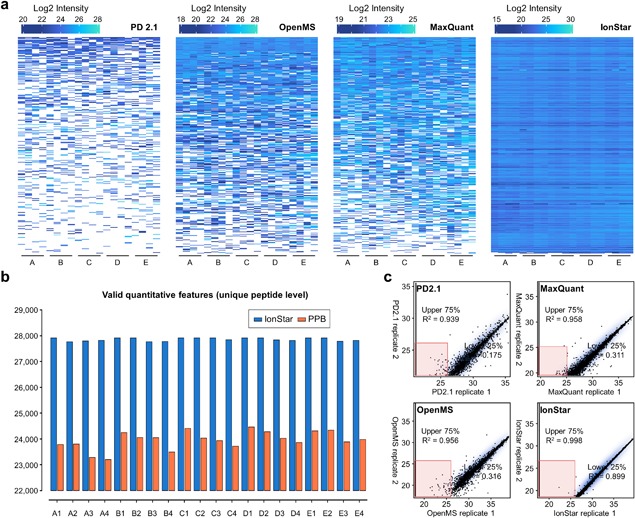
The rapidly-advancing field of pharmaceutical and clinical research calls for systematic, molecular-level characterization of complex biological systems. To this end, quantitative proteomics represents a powerful tool but an optimal solution for reliable large-cohort proteomics analysis, as frequently involved in pharmaceutical/clinical investigations, is urgently needed. Large-cohort analysis remains challenging owing to the deteriorating quantitative quality and snowballing missing data and false-positive discovery of altered proteins when sample size increases. MS1 ion current-based methods, which have become an important class of label-free quantification techniques during the past decade, show considerable potential to achieve reproducible protein measurements in large cohorts with high quantitative accuracy/precision. Nonetheless, in order to fully unleash this potential, several critical prerequisites should be met. Here we provide an overview of the rationale of MS1-based strategies and then important considerations for experimental and data processing techniques, with the emphasis on (i) efficient and reproducible sample preparation and LC separation; (ii) sensitive, selective and high-resolution MS detection; iii)accurate chromatographic alignment; (iv) sensitive and selective generation of quantitative features; and (v) optimal post-feature-generation data quality control. Prominent technical developments in these aspects are discussed. Finally, we reviewed applications of MS1-based strategy in disease mechanism studies, biomarker discovery, and pharmaceutical investigations.
Keywords: LC-MS; MS1 quantification; ion current-based proteomics; large cohorts; reproducible protein measurement.
© 2019 The Authors. Mass Spectrometry Reviews Published by Wiley Periodicals, Inc.
Figures
References
-
- Allet N, Barrillat N, Baussant T, et al. 2004. In vitro and in silico processes to identify differentially expressed proteins. Proteomics 4(8):2333–2351. - PubMed
-
- An B, Zhang M, Johnson RW, Qu J. 2015. Surfactant‐aided precipitation/on‐pellet‐digestion (SOD) procedure provides robust and rapid sample preparation for reproducible, accurate and sensitive LC/MS quantification of therapeutic protein in plasma and tissues. Anal Chem 87(7):4023–4029. - PubMed
-
- Andreev VP, Rejtar T, Chen HS, Moskovets EV, Ivanov AR, Karger BL. 2003. A universal denoising and peak picking algorithm for LC‐MS based on matched filtration in the chromatographic time domain. Anal Chem 75(22):6314–6326. - PubMed
-
- Ballardini R, Benevento M, Arrigoni G, Pattini L, Roda A. 2011. Mass untangler: A novel alignment tool for label‐free liquid chromatography‐mass spectrometry proteomic data. J Chromatogr A 1218(49):8859–8868. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
