

Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation
- PMID: 30921319
- PMCID: PMC6469801
- DOI: 10.1371/journal.pbio.2006859
Prophylactic TLR9 stimulation reduces brain metastasis through microglia activation
Abstract
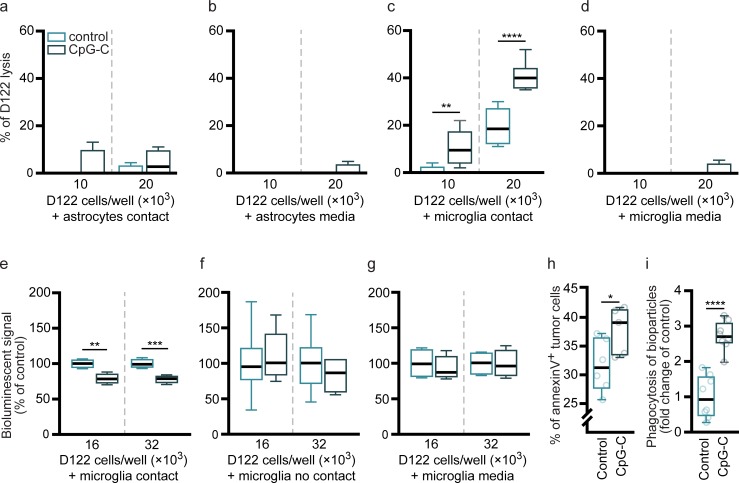
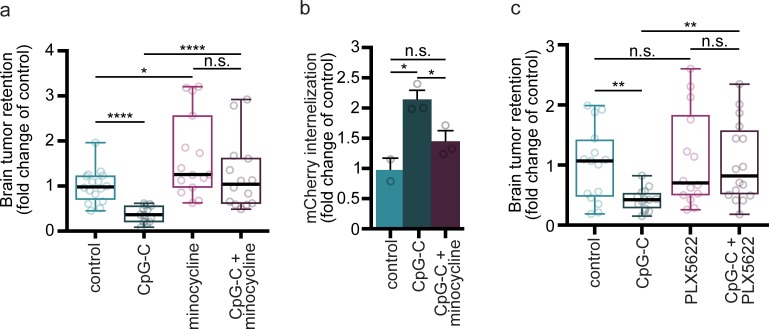
Brain metastases are prevalent in various types of cancer and are often terminal, given the low efficacy of available therapies. Therefore, preventing them is of utmost clinical relevance, and prophylactic treatments are perhaps the most efficient strategy. Here, we show that systemic prophylactic administration of a toll-like receptor (TLR) 9 agonist, CpG-C, is effective against brain metastases. Acute and chronic systemic administration of CpG-C reduced tumor cell seeding and growth in the brain in three tumor models in mice, including metastasis of human and mouse lung cancer, and spontaneous melanoma-derived brain metastasis. Studying mechanisms underlying the therapeutic effects of CpG-C, we found that in the brain, unlike in the periphery, natural killer (NK) cells and monocytes are not involved in controlling metastasis. Next, we demonstrated that the systemically administered CpG-C is taken up by endothelial cells, astrocytes, and microglia, without affecting blood-brain barrier (BBB) integrity and tumor brain extravasation. In vitro assays pointed to microglia, but not astrocytes, as mediators of CpG- C effects through increased tumor killing and phagocytosis, mediated by direct microglia-tumor contact. In vivo, CpG-C-activated microglia displayed elevated mRNA expression levels of apoptosis-inducing and phagocytosis-related genes. Intravital imaging showed that CpG-C-activated microglia cells contact, kill, and phagocytize tumor cells in the early stages of tumor brain invasion more than nonactivated microglia. Blocking in vivo activation of microglia with minocycline, and depletion of microglia with a colony-stimulating factor 1 inhibitor, indicated that microglia mediate the antitumor effects of CpG-C. Overall, the results suggest prophylactic CpG-C treatment as a new intervention against brain metastasis, through an essential activation of microglia.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures







Comment in
-
Better safe than sorry: a potential prophylactic treatment for brain metastasis.Nat Rev Cancer. 2019 Jun;19(6):303. doi: 10.1038/s41568-019-0150-8. Nat Rev Cancer. 2019. PMID: 31053803 No abstract available.
References
-
- Kim S-WS, Kim J-S, Park ES, Lee J-S, Lin Q, Langley RR, et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia [Internet]. 2011. March [cited 2017 Nov 3];13(3). Available from: http://www.ncbi.nlm.nih.gov/pubmed/21390191 - PMC - PubMed
-
- Kienast Y, Winkler F. Therapy and prophylaxis of brain metastases. Expert Rev Anticancer Ther [Internet]. 2010. November;10(11):1763–77 [cited 2019 Mar 20]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21080803 10.1586/era.10.165 - DOI - PubMed
-
- Kodack DP, Askoxylakis V, Ferraro GB, Fukumura D, Jain RK. Emerging Strategies for Treating Brain Metastases from Breast Cancer. Cancer Cell [Internet]. 2015. February 9 [cited 2018 Mar 3];27(2):163–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25670078 10.1016/j.ccell.2015.01.001 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
