

Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers
- PMID: 30921324
- PMCID: PMC6438456
- DOI: 10.1371/journal.pcbi.1006658
Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers
Erratum in
-
Correction: Review: Precision medicine and driver mutations: Computational methods, functional assays and conformational principles for interpreting cancer drivers.PLoS Comput Biol. 2019 Jun 12;15(6):e1007114. doi: 10.1371/journal.pcbi.1007114. eCollection 2019 Jun. PLoS Comput Biol. 2019. PMID: 31188819 Free PMC article.
Abstract
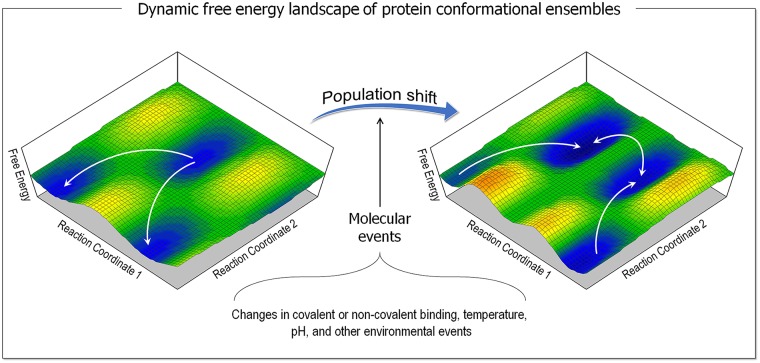
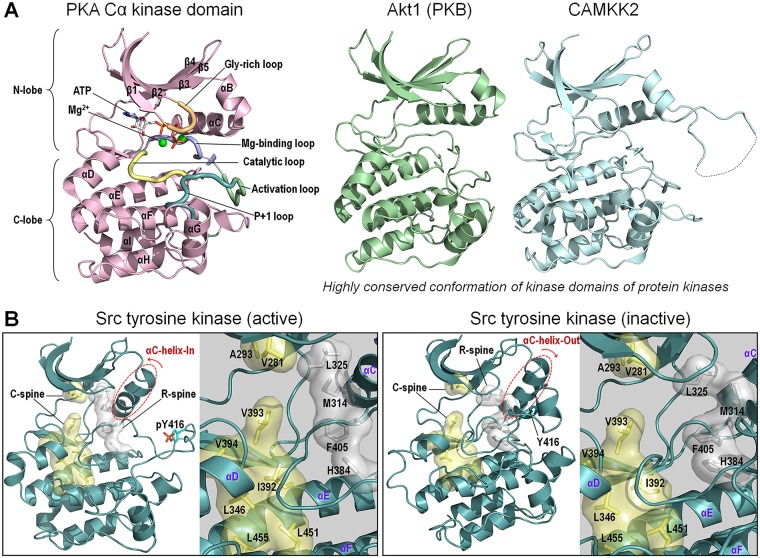
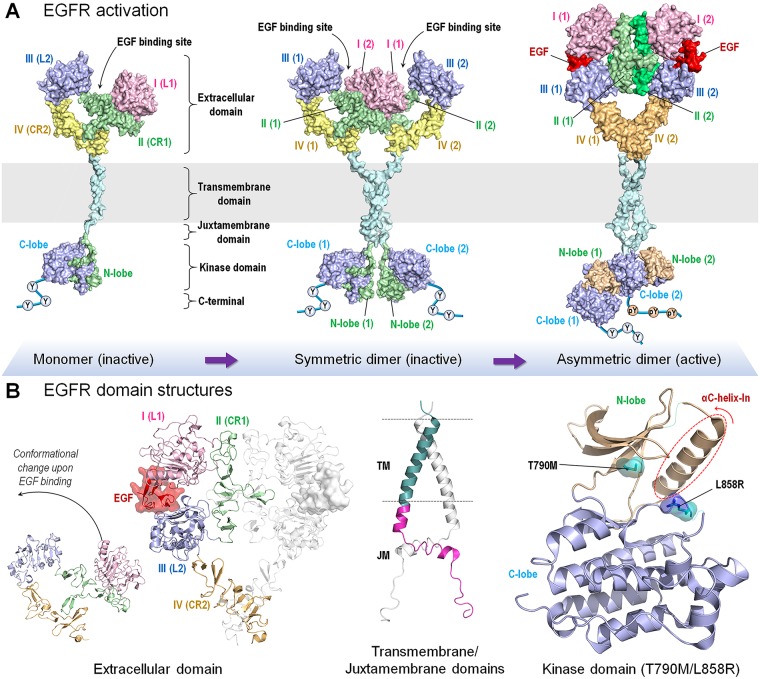
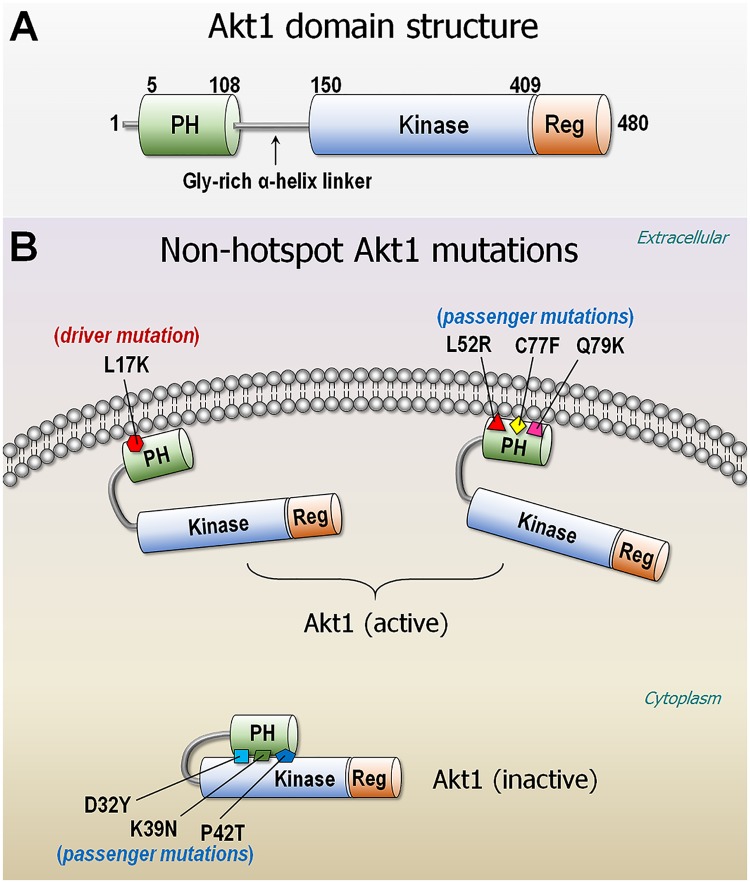
At the root of the so-called precision medicine or precision oncology, which is our focus here, is the hypothesis that cancer treatment would be considerably better if therapies were guided by a tumor's genomic alterations. This hypothesis has sparked major initiatives focusing on whole-genome and/or exome sequencing, creation of large databases, and developing tools for their statistical analyses-all aspiring to identify actionable alterations, and thus molecular targets, in a patient. At the center of the massive amount of collected sequence data is their interpretations that largely rest on statistical analysis and phenotypic observations. Statistics is vital, because it guides identification of cancer-driving alterations. However, statistics of mutations do not identify a change in protein conformation; therefore, it may not define sufficiently accurate actionable mutations, neglecting those that are rare. Among the many thematic overviews of precision oncology, this review innovates by further comprehensively including precision pharmacology, and within this framework, articulating its protein structural landscape and consequences to cellular signaling pathways. It provides the underlying physicochemical basis, thereby also opening the door to a broader community.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
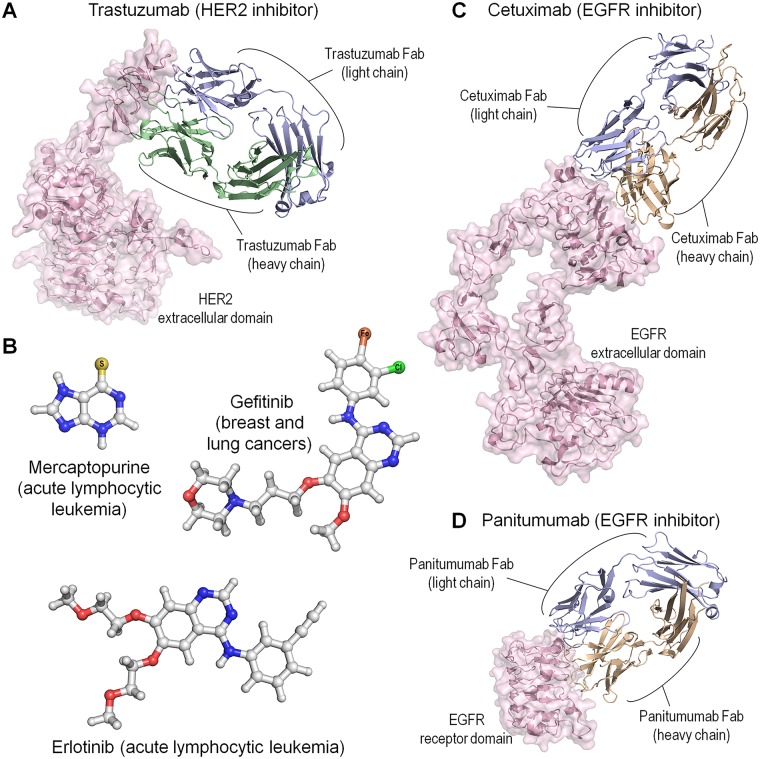
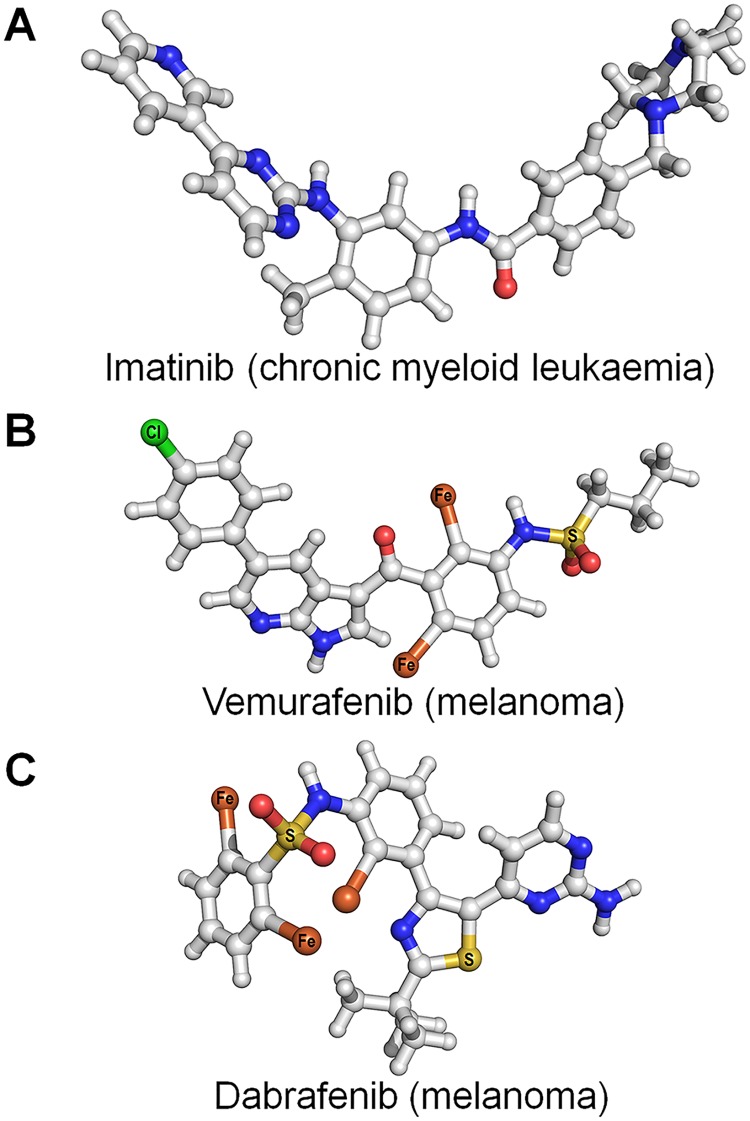
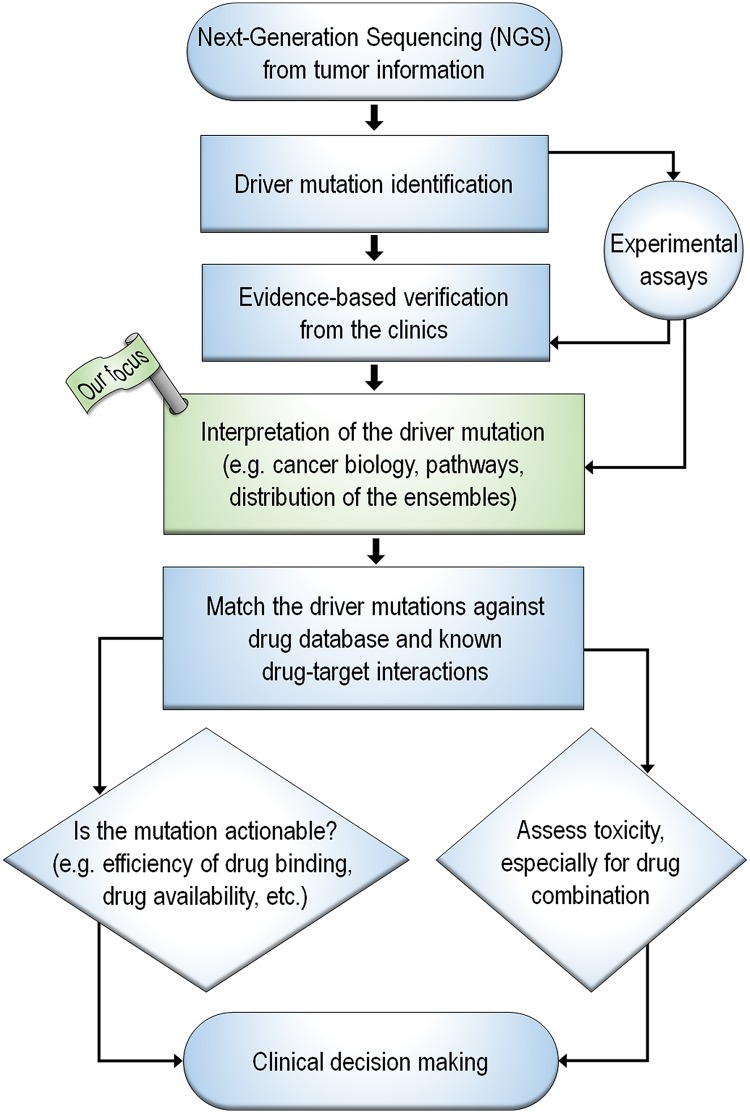
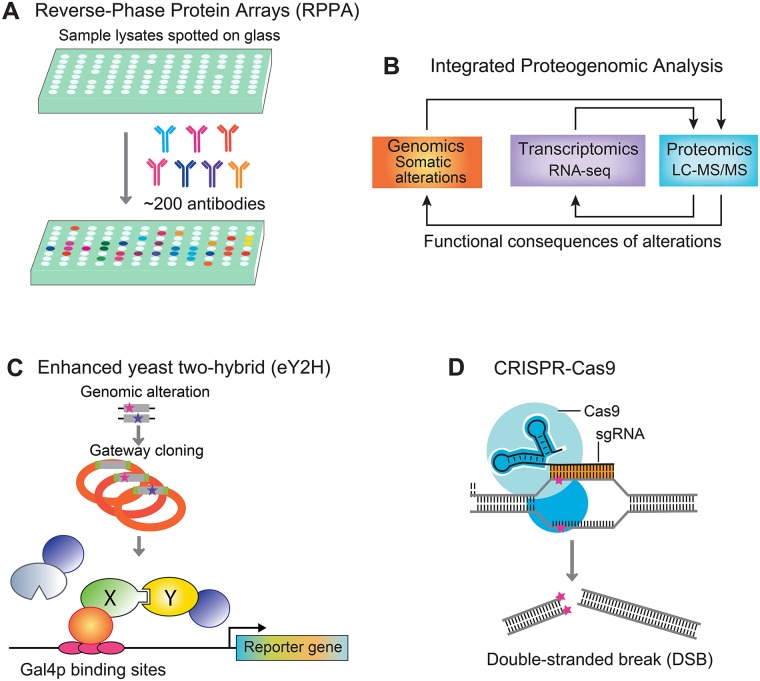

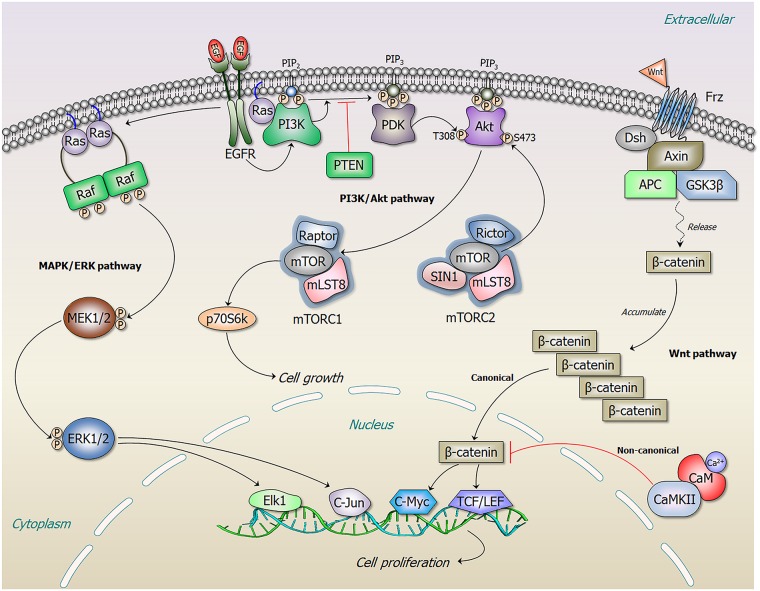
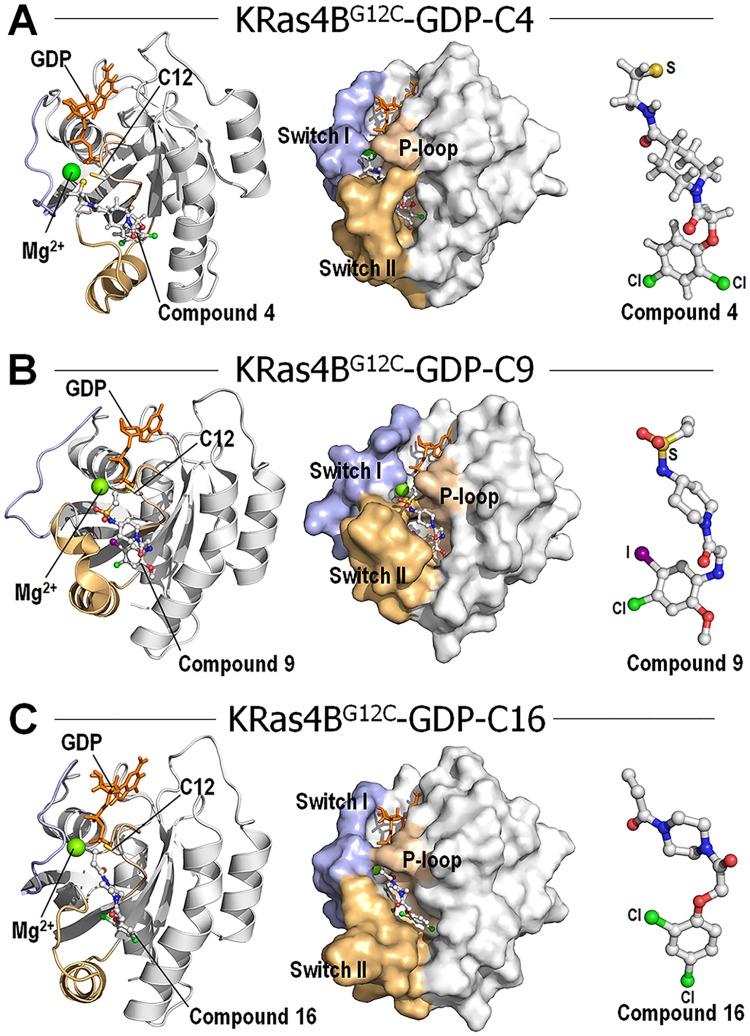
References
-
- Manem VSK, Salgado R, Aftimos P, Sotiriou C, Haibe-Kains B (2017) Network science in clinical trials: A patient-centered approach. Semin Cancer Biol. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
