

Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling
- PMID: 30922063
- PMCID: PMC6553980
- DOI: 10.1161/CIRCULATIONAHA.118.036099
Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling
Abstract
Background: Heart failure, which is a major global health problem, is often preceded by pathological cardiac hypertrophy. The expansion of the cardiac vasculature, to maintain adequate supply of oxygen and nutrients, is a key determinant of whether the heart grows in a physiological compensated manner or a pathological decompensated manner. Bidirectional endothelial cell (EC)-cardiomyocyte (CMC) cross talk via cardiokine and angiocrine signaling plays an essential role in the regulation of cardiac growth and homeostasis. Currently, the mechanisms involved in the EC-CMC interaction are not fully understood, and very little is known about the EC-derived signals involved. Understanding how an excess of angiogenesis induces cardiac hypertrophy and how ECs regulate CMC homeostasis could provide novel therapeutic targets for heart failure.
Methods: Genetic mouse models were used to delete vascular endothelial growth factor (VEGF) receptors, adeno-associated viral vectors to transduce the myocardium, and pharmacological inhibitors to block VEGF and ErbB signaling in vivo. Cell culture experiments were used for mechanistic studies, and quantitative polymerase chain reaction, microarrays, ELISA, and immunohistochemistry were used to analyze the cardiac phenotypes.
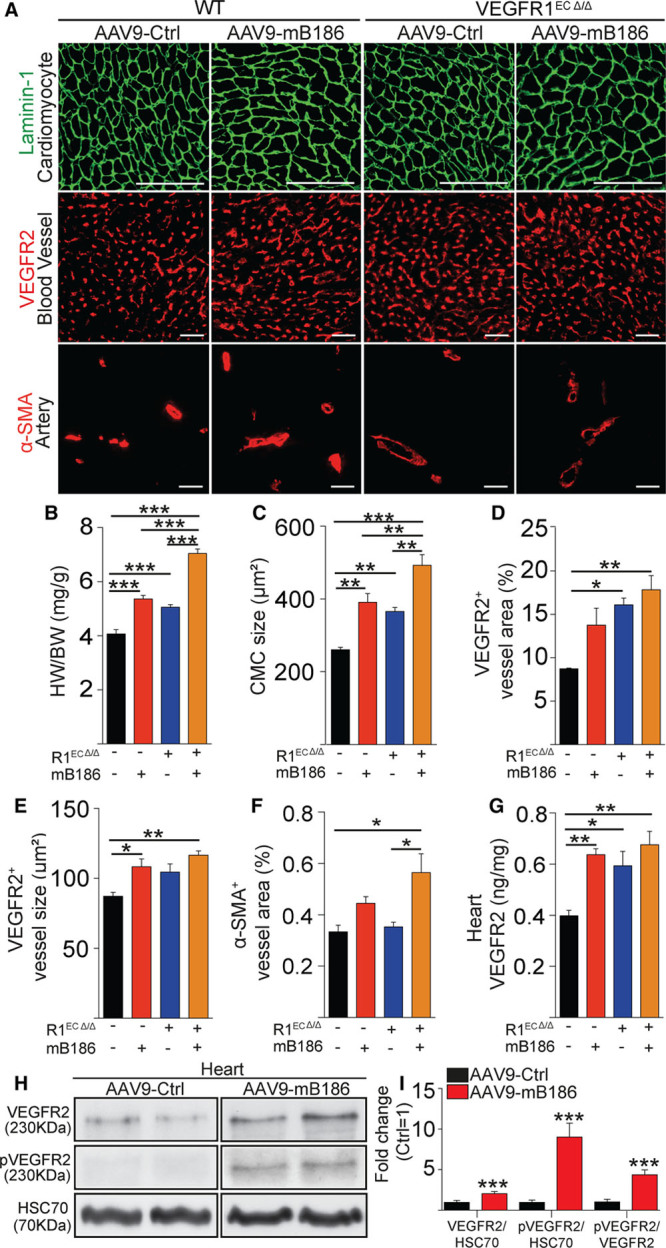
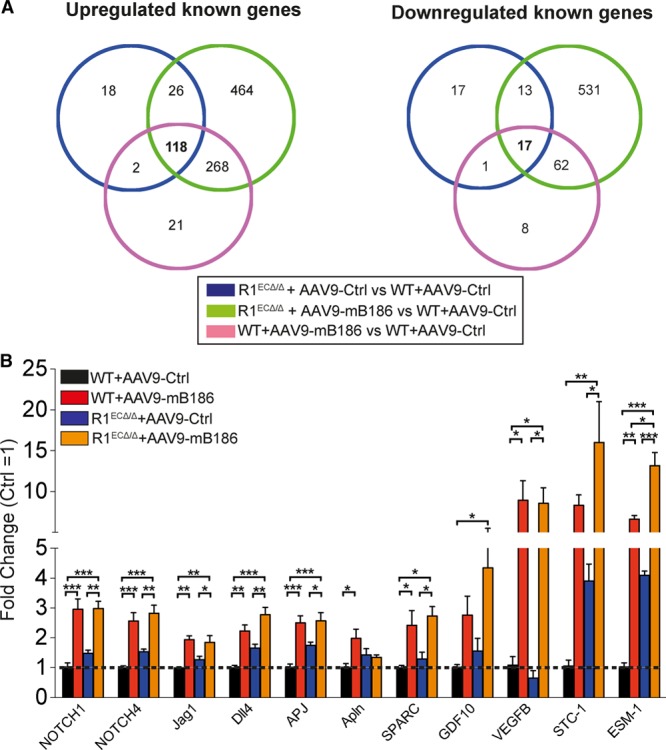
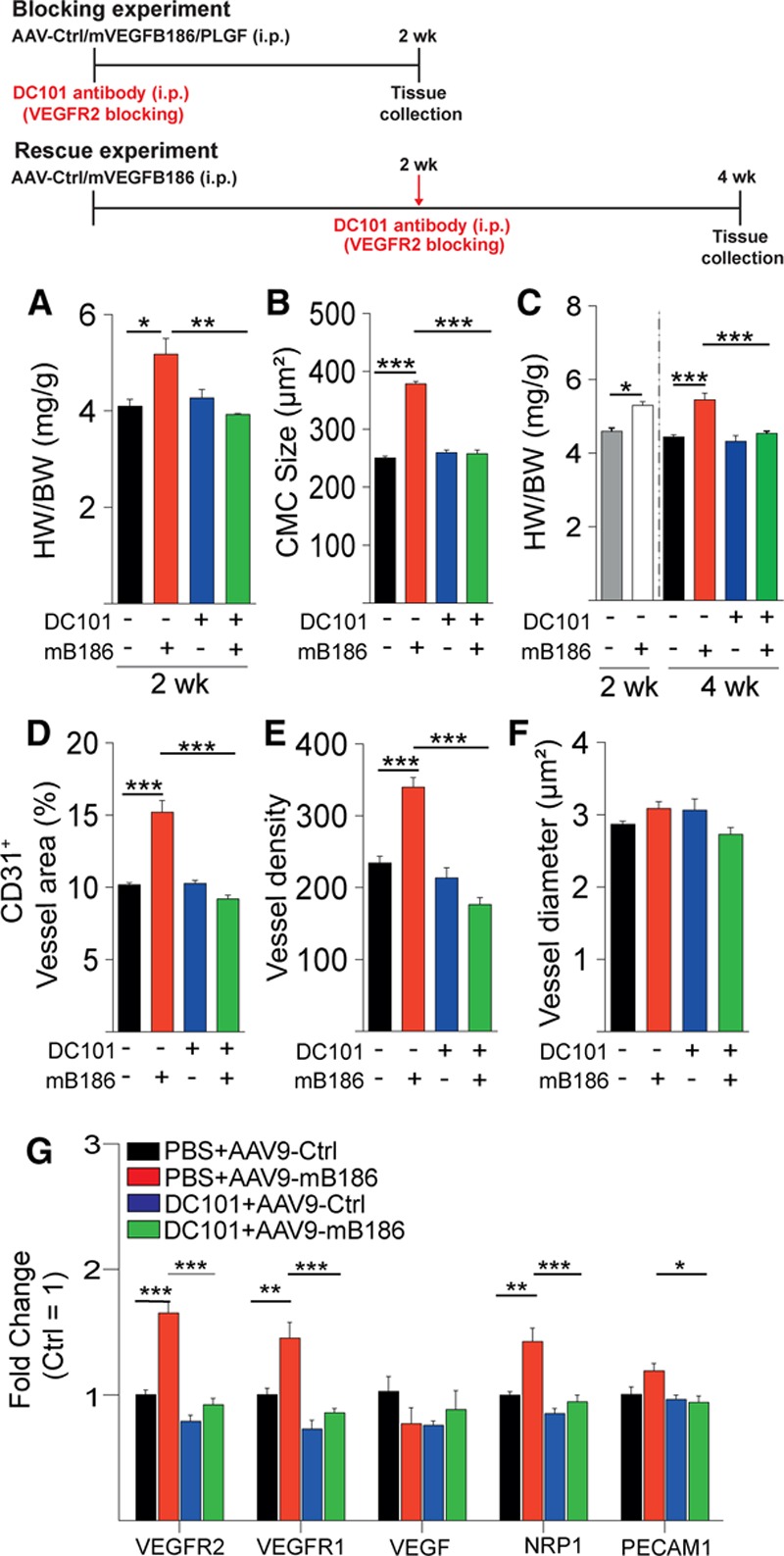
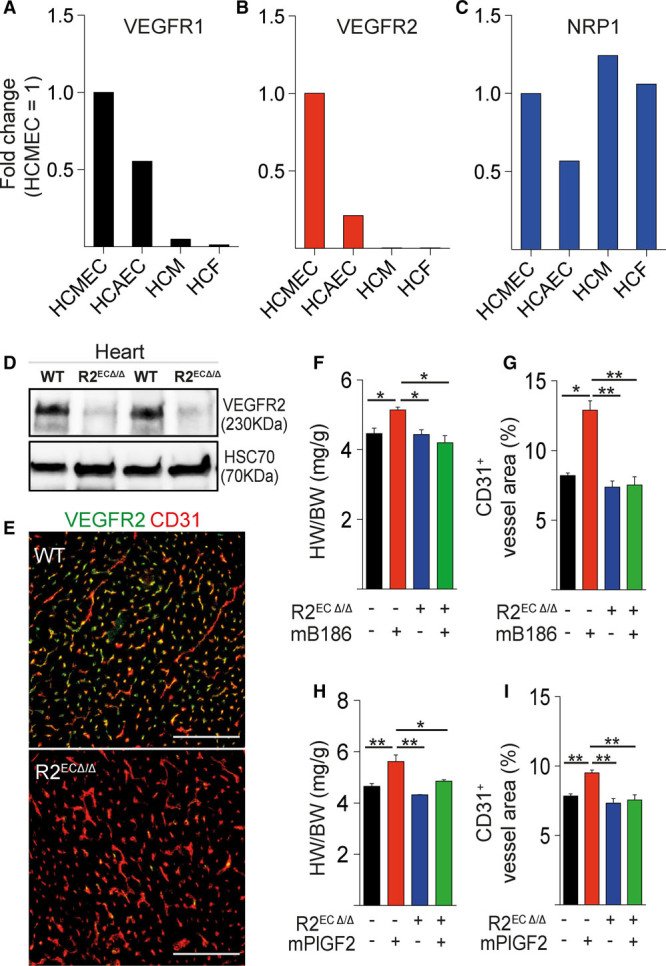
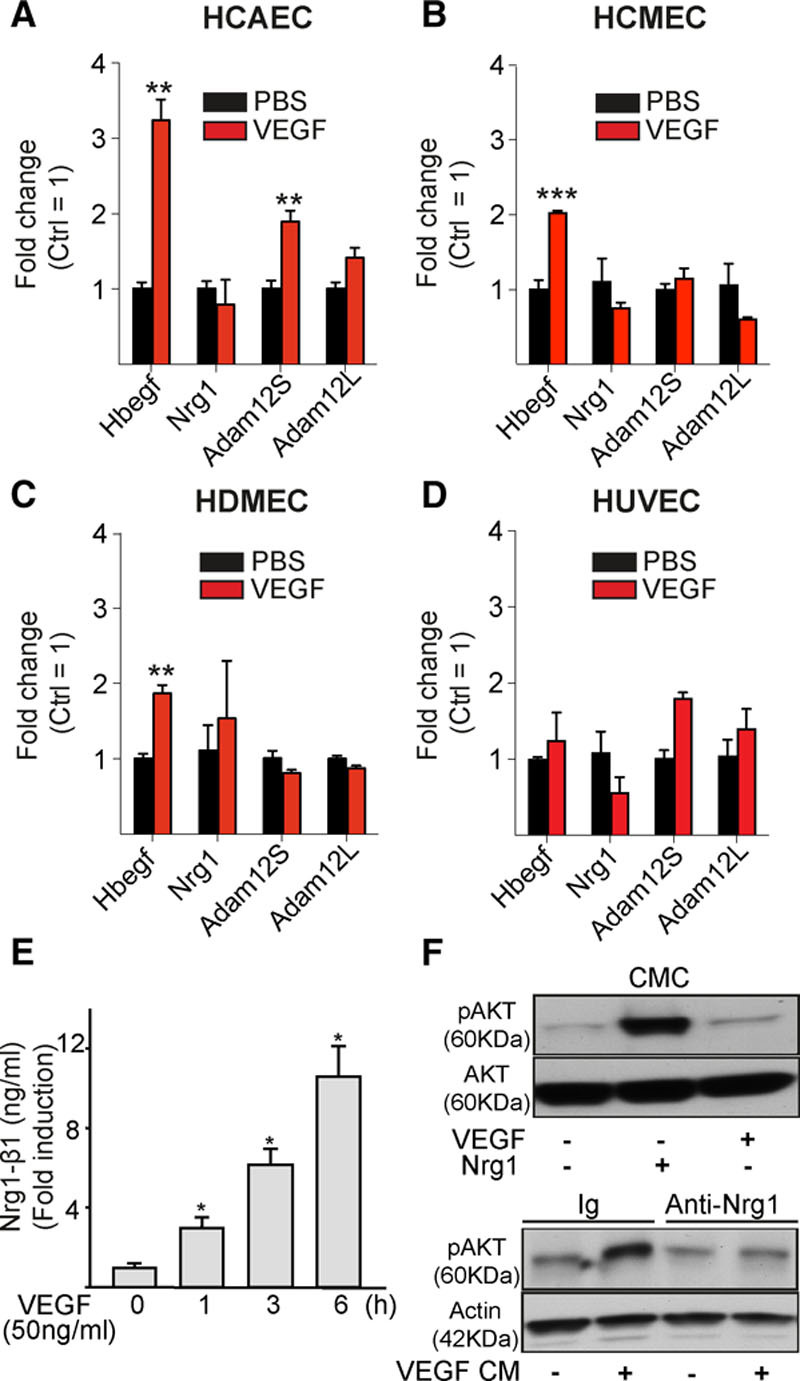
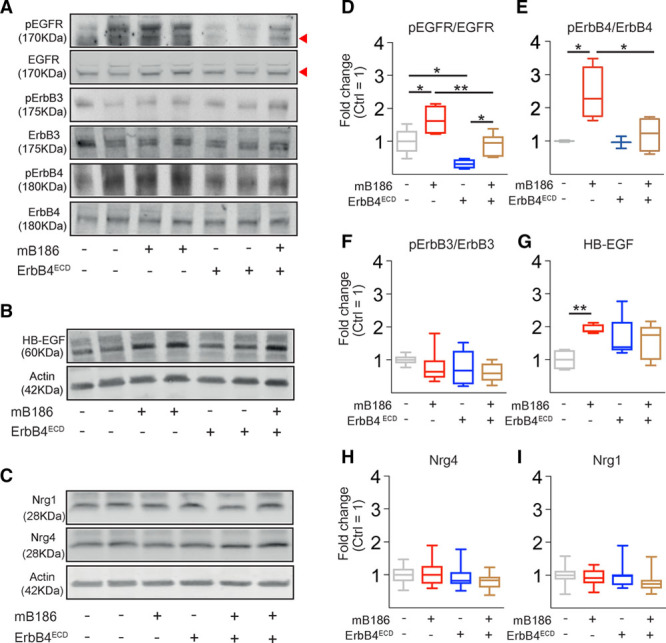
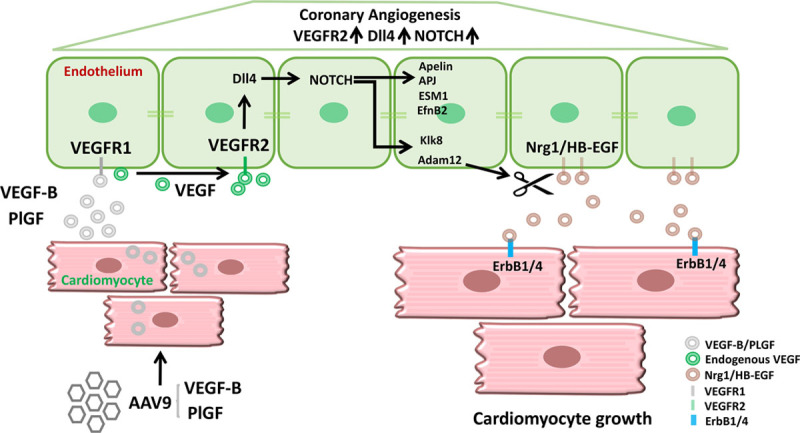
Results: Both EC deletion of VEGF receptor (VEGFR)-1 and adeno-associated viral vector-mediated delivery of the VEGFR1-specific ligands VEGF-B or placental growth factor into the myocardium increased the coronary vasculature and induced CMC hypertrophy in adult mice. The resulting cardiac hypertrophy was physiological, as indicated by preserved cardiac function and exercise capacity and lack of pathological gene activation. These changes were mediated by increased VEGF signaling via endothelial VEGFR2, because the effects of VEGF-B and placental growth factor on both angiogenesis and CMC growth were fully inhibited by treatment with antibodies blocking VEGFR2 or by endothelial deletion of VEGFR2. To identify activated pathways downstream of VEGFR2, whole-genome transcriptomics and secretome analyses were performed, and the Notch and ErbB pathways were shown to be involved in transducing signals for EC-CMC cross talk in response to angiogenesis. Pharmacological or genetic blocking of ErbB signaling also inhibited part of the VEGF-B-induced effects in the heart.
Conclusions: This study reveals that cross talk between the EC VEGFR2 and CMC ErbB signaling pathways coordinates CMC hypertrophy with angiogenesis, contributing to physiological cardiac growth.
Keywords: HB-EGF; VEGF-B; VEGFR-1; angiogenesis; hypertrophy.
Figures

Comment in
-
Endothelial Cell-Derived Angiocrines Elicit Physiological Cardiomyocyte Hypertrophy.Circulation. 2019 May 28;139(22):2585-2587. doi: 10.1161/CIRCULATIONAHA.119.040632. Epub 2019 May 28. Circulation. 2019. PMID: 31136219 No abstract available.
References
-
- Hudlicka O, Brown M, Egginton S. Angiogenesis in skeletal and cardiac muscle. Physiol Rev. 1992;72:369–417. doi: 10.1152/physrev.1992.72.2.369. - PubMed
-
- Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. doi: 10.1016/j.yjmcc.2016.06.001. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
