

The PI3K and MAPK/p38 pathways control stress granule assembly in a hierarchical manner
- PMID: 30923191
- PMCID: PMC6441495
- DOI: 10.26508/lsa.201800257
The PI3K and MAPK/p38 pathways control stress granule assembly in a hierarchical manner
Abstract
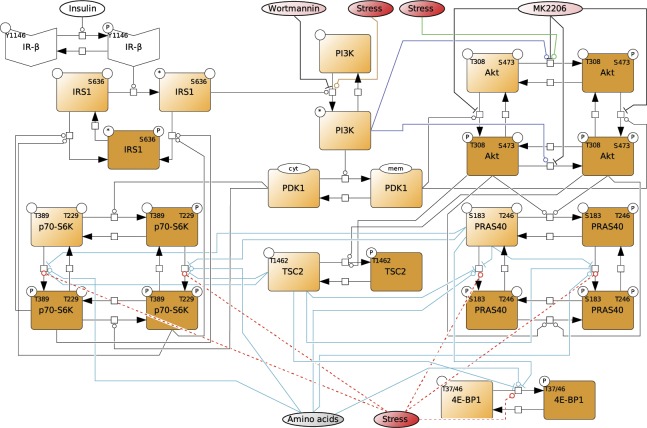
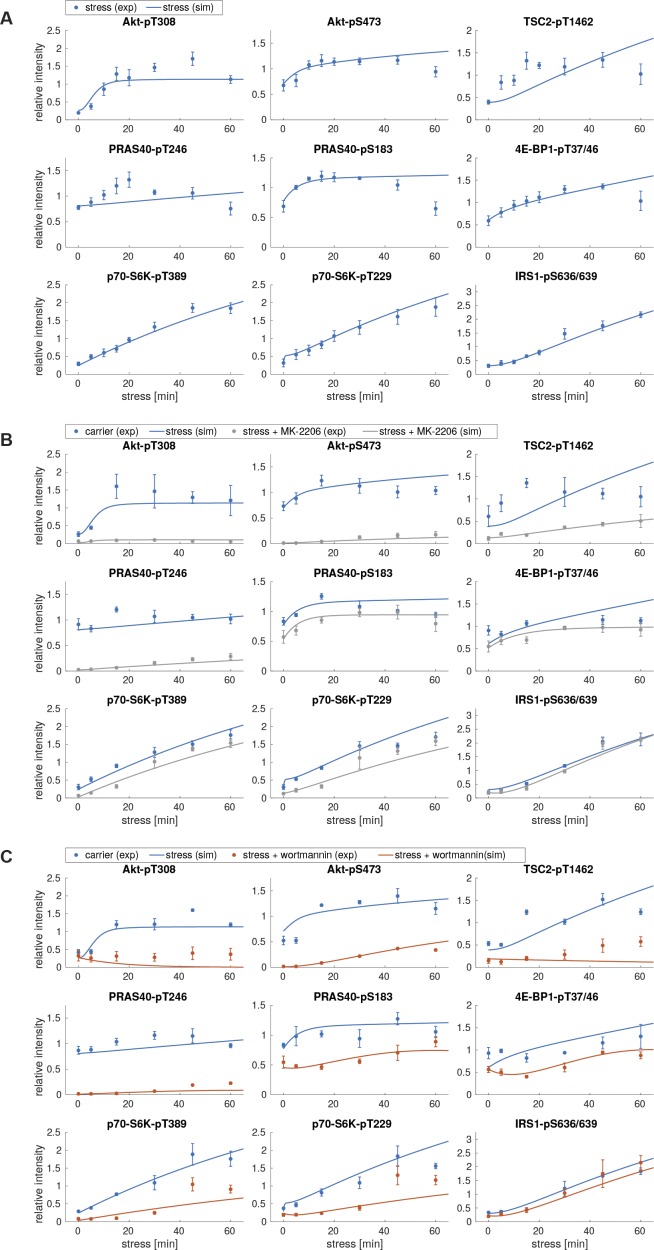
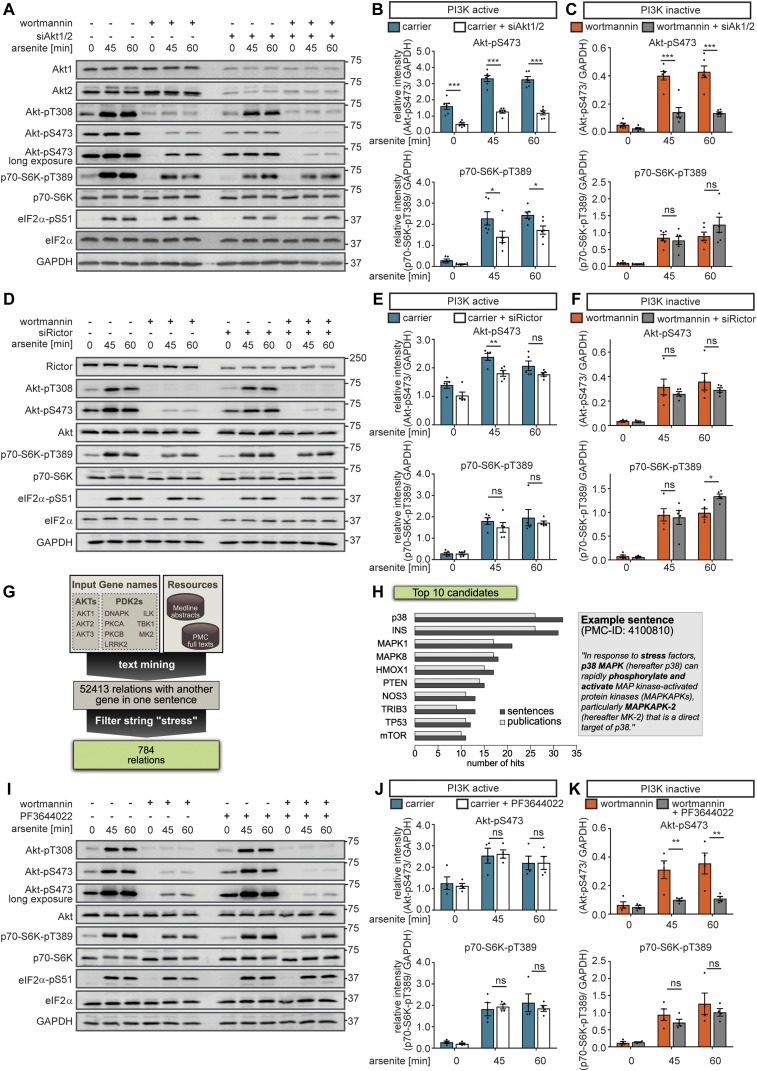
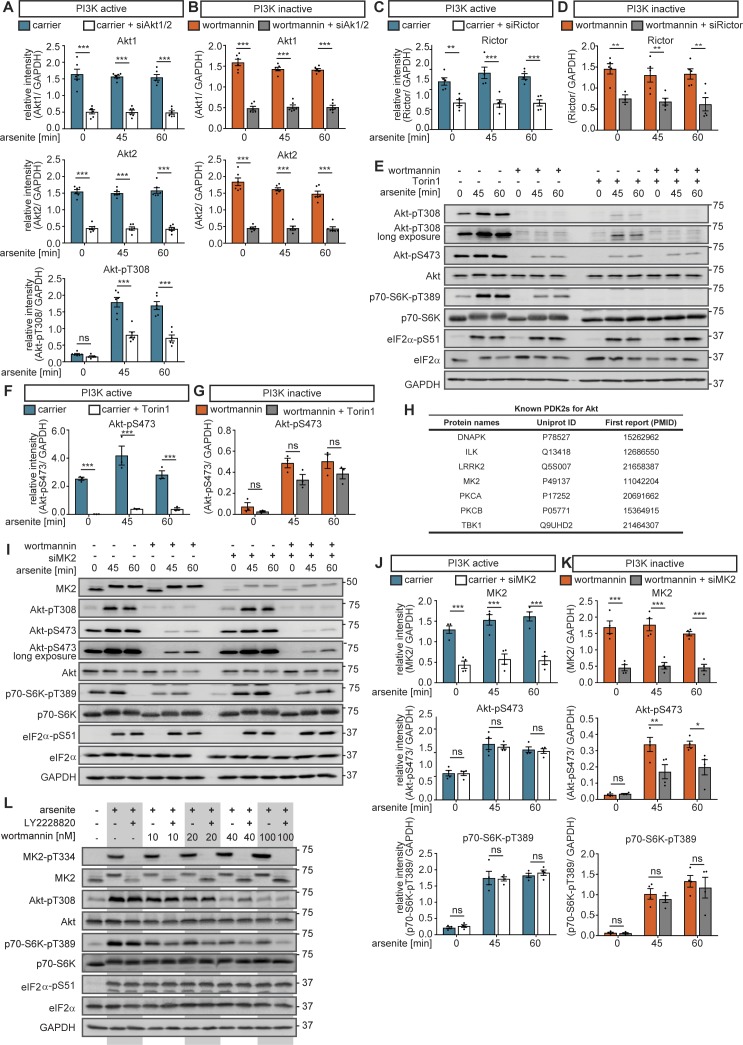
All cells and organisms exhibit stress-coping mechanisms to ensure survival. Cytoplasmic protein-RNA assemblies termed stress granules are increasingly recognized to promote cellular survival under stress. Thus, they might represent tumor vulnerabilities that are currently poorly explored. The translation-inhibitory eIF2α kinases are established as main drivers of stress granule assembly. Using a systems approach, we identify the translation enhancers PI3K and MAPK/p38 as pro-stress-granule-kinases. They act through the metabolic master regulator mammalian target of rapamycin complex 1 (mTORC1) to promote stress granule assembly. When highly active, PI3K is the main driver of stress granules; however, the impact of p38 becomes apparent as PI3K activity declines. PI3K and p38 thus act in a hierarchical manner to drive mTORC1 activity and stress granule assembly. Of note, this signaling hierarchy is also present in human breast cancer tissue. Importantly, only the recognition of the PI3K-p38 hierarchy under stress enabled the discovery of p38's role in stress granule formation. In summary, we assign a new pro-survival function to the key oncogenic kinases PI3K and p38, as they hierarchically promote stress granule formation.
© 2019 Heberle et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
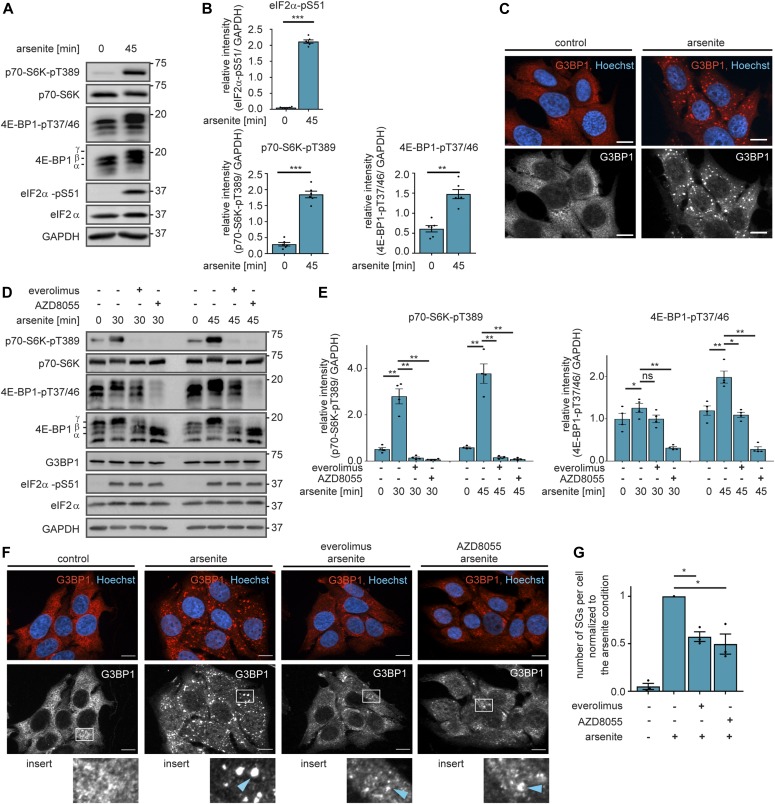
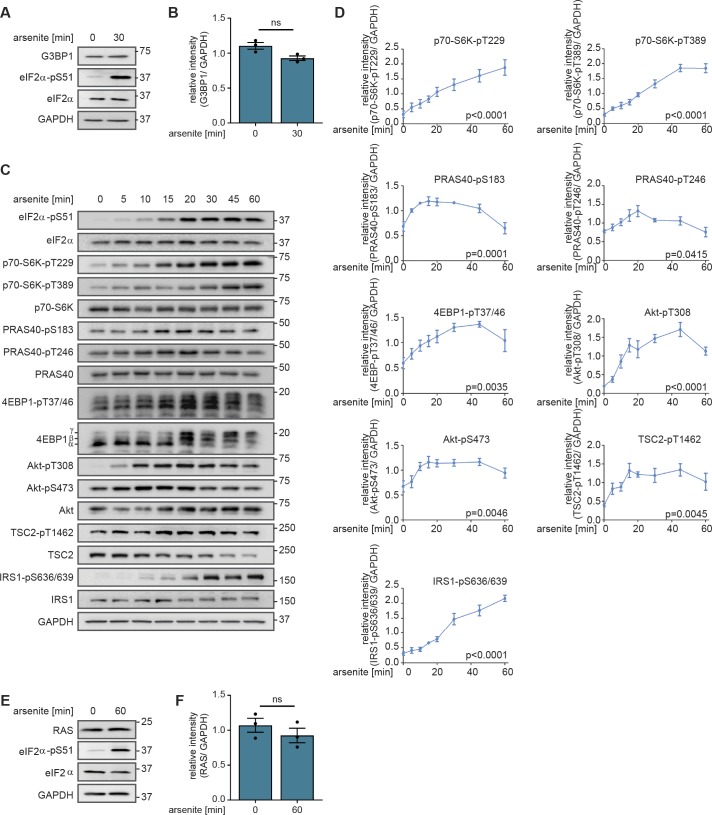
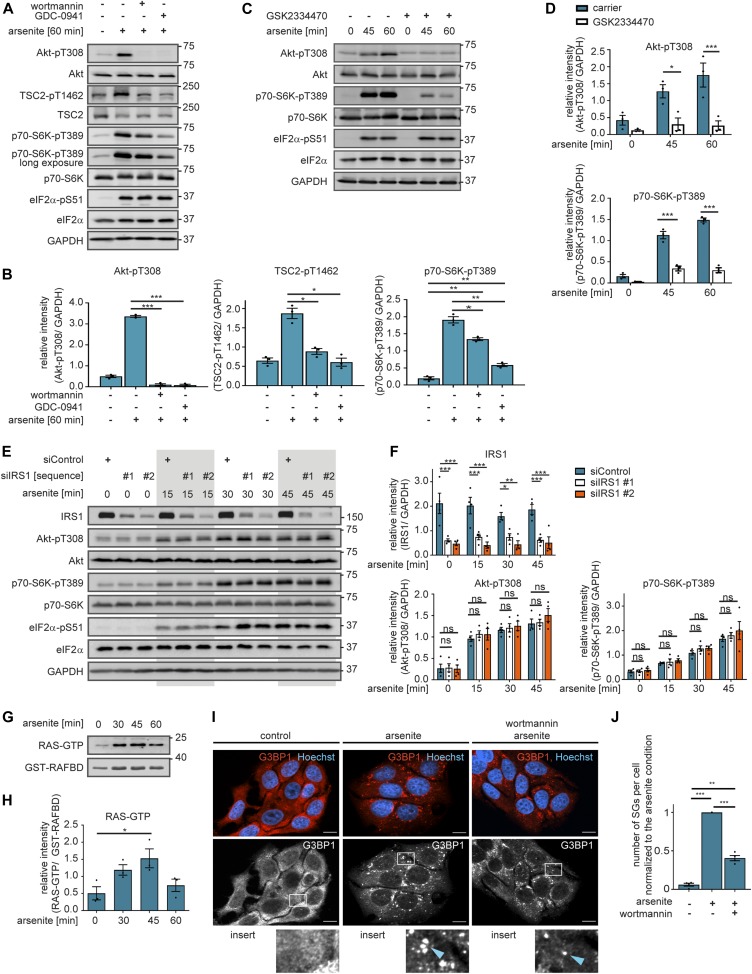
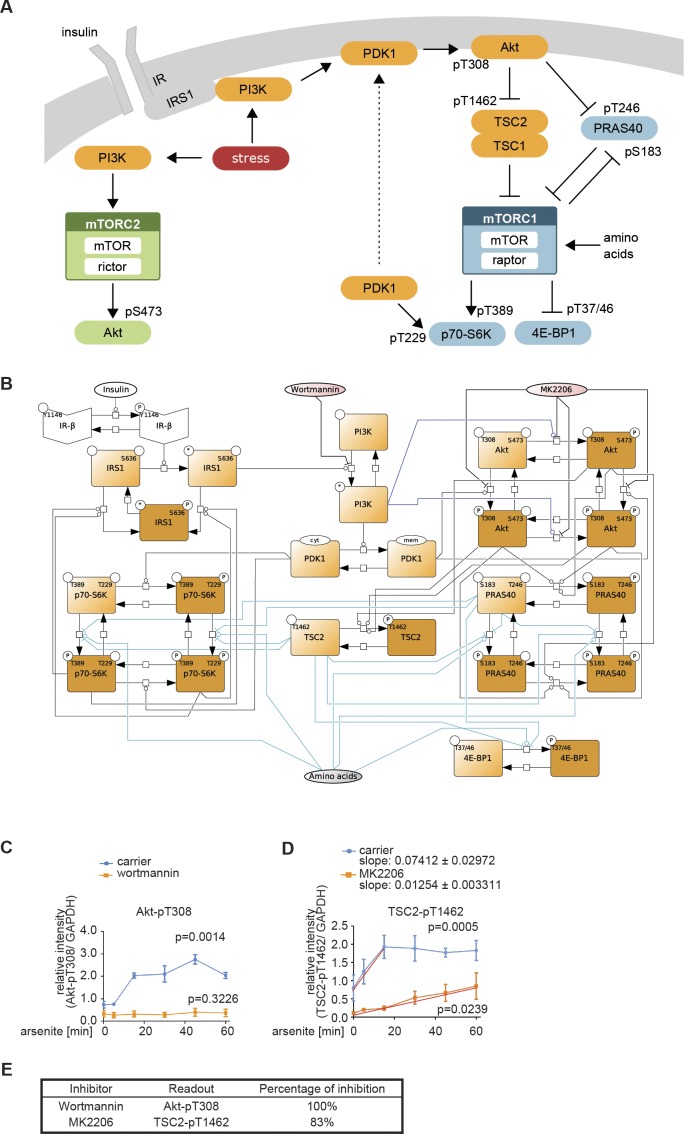
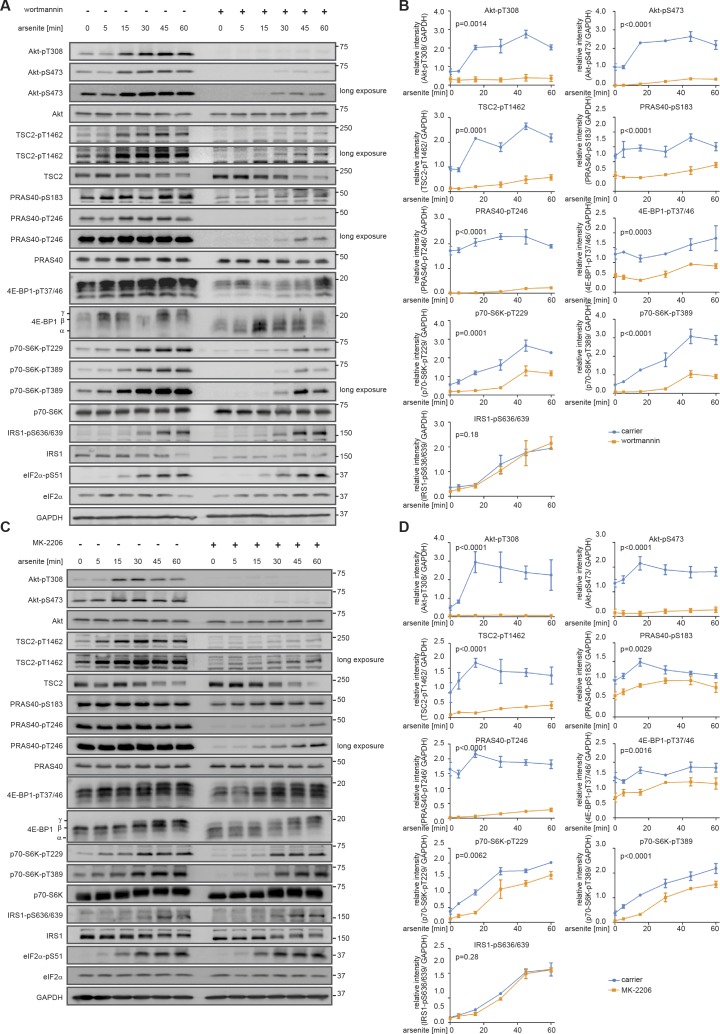
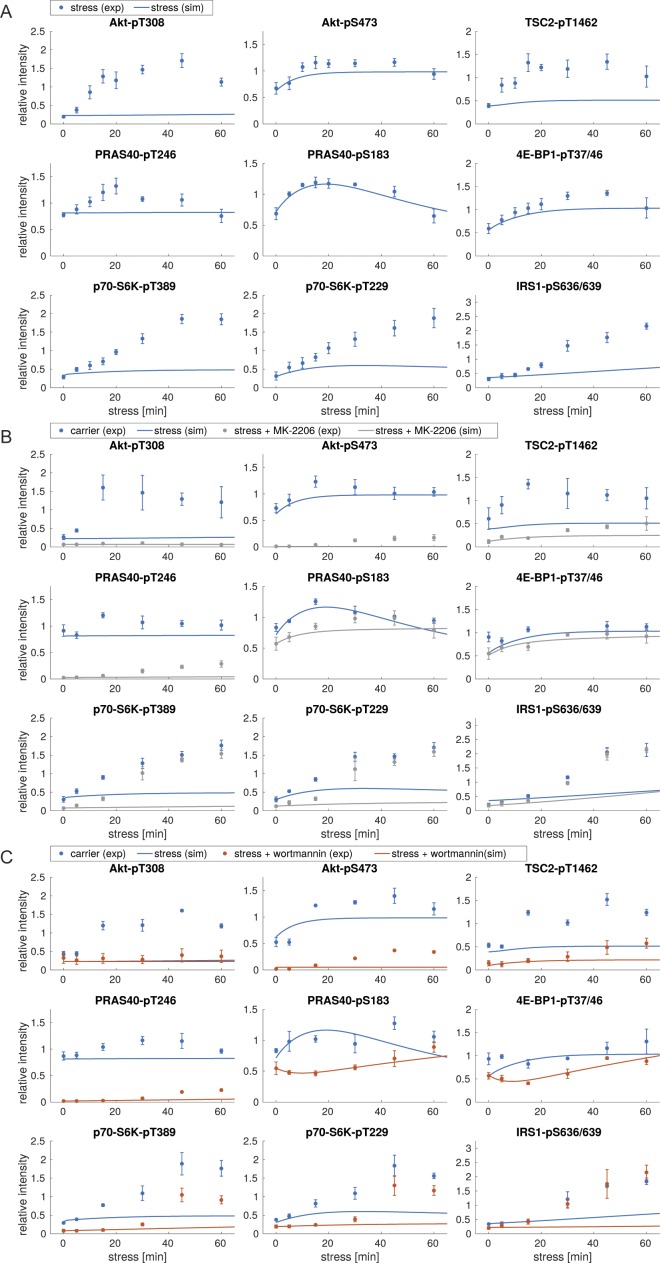
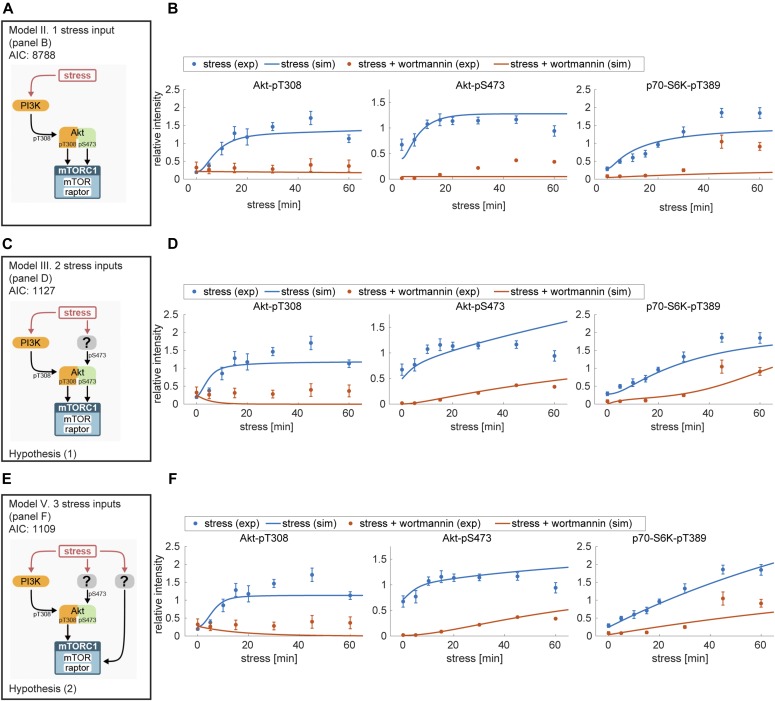
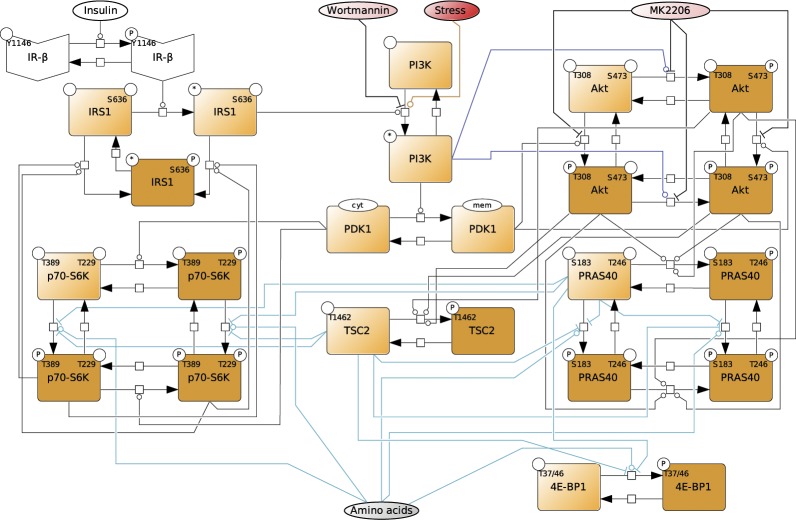
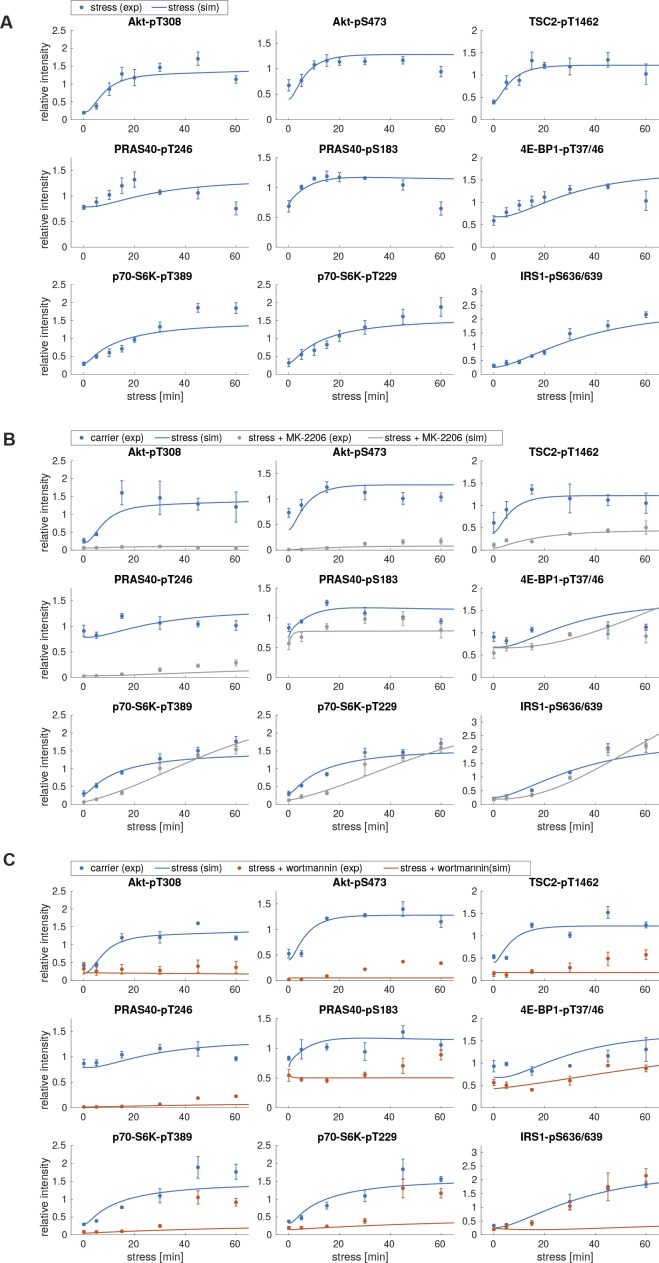
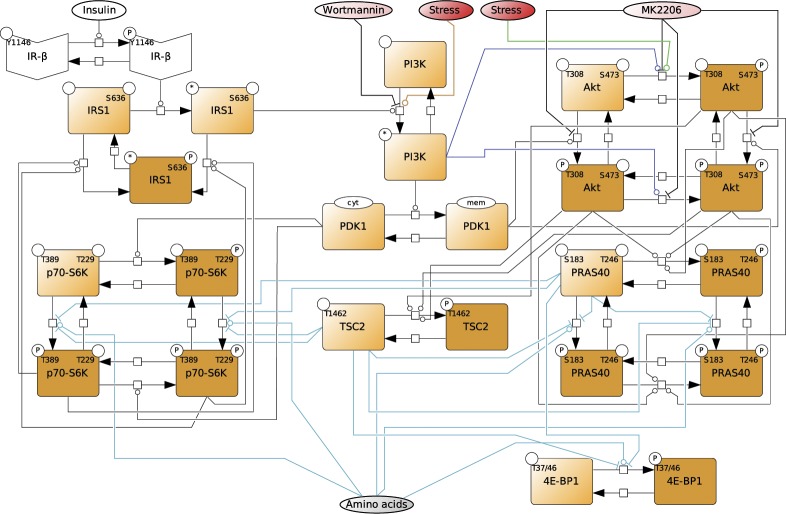
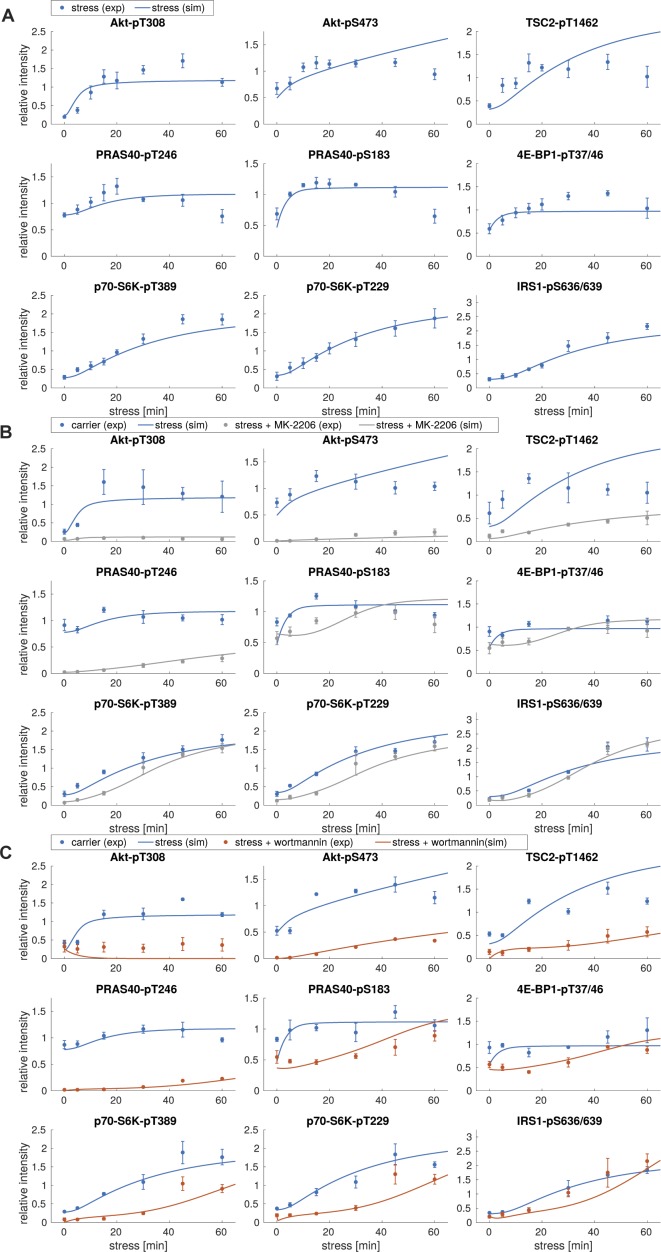
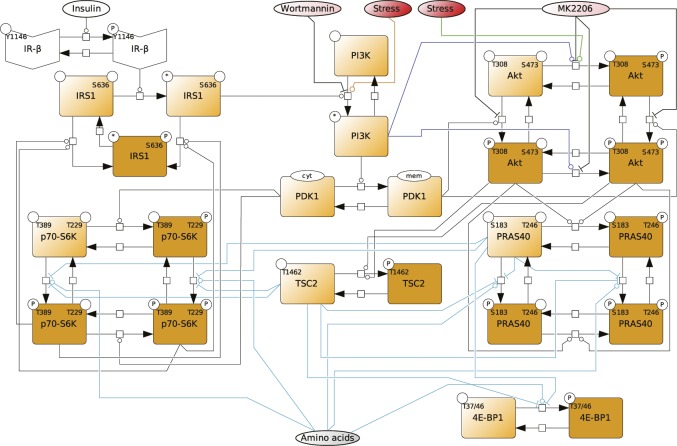

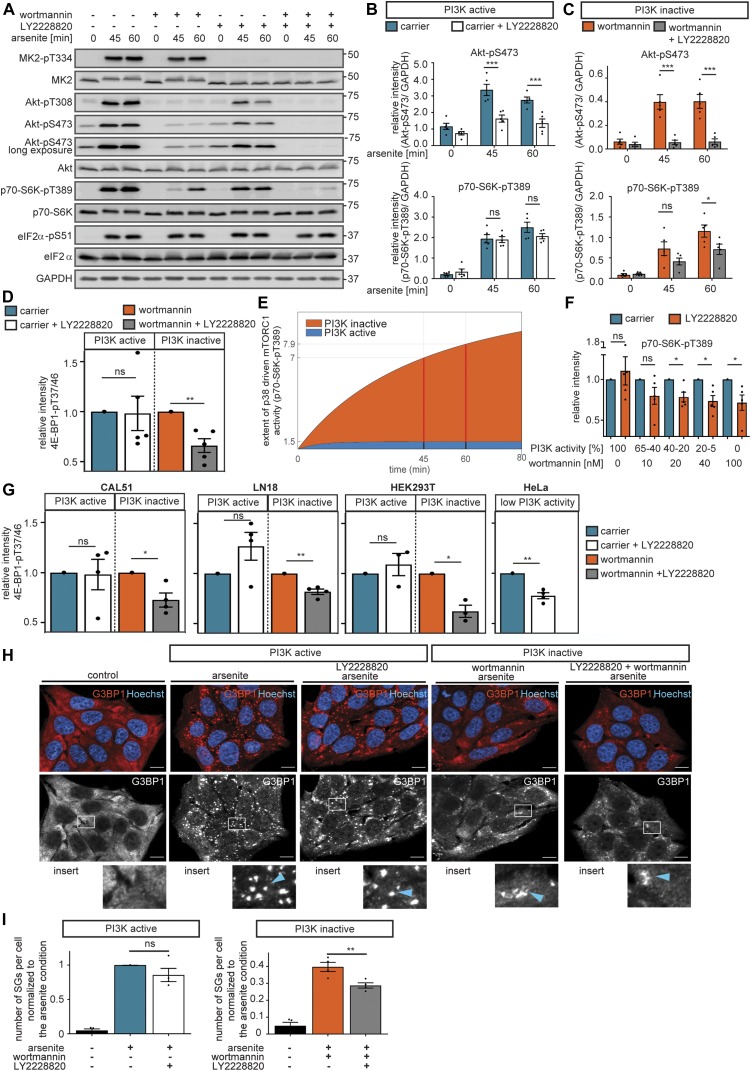
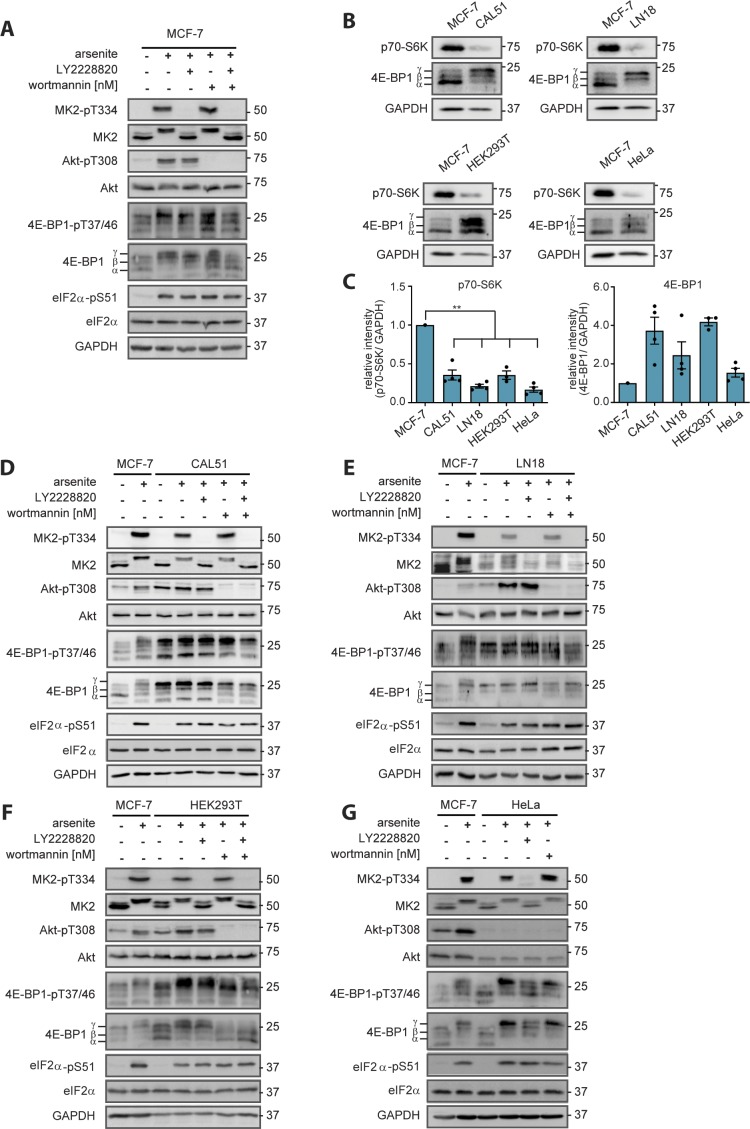
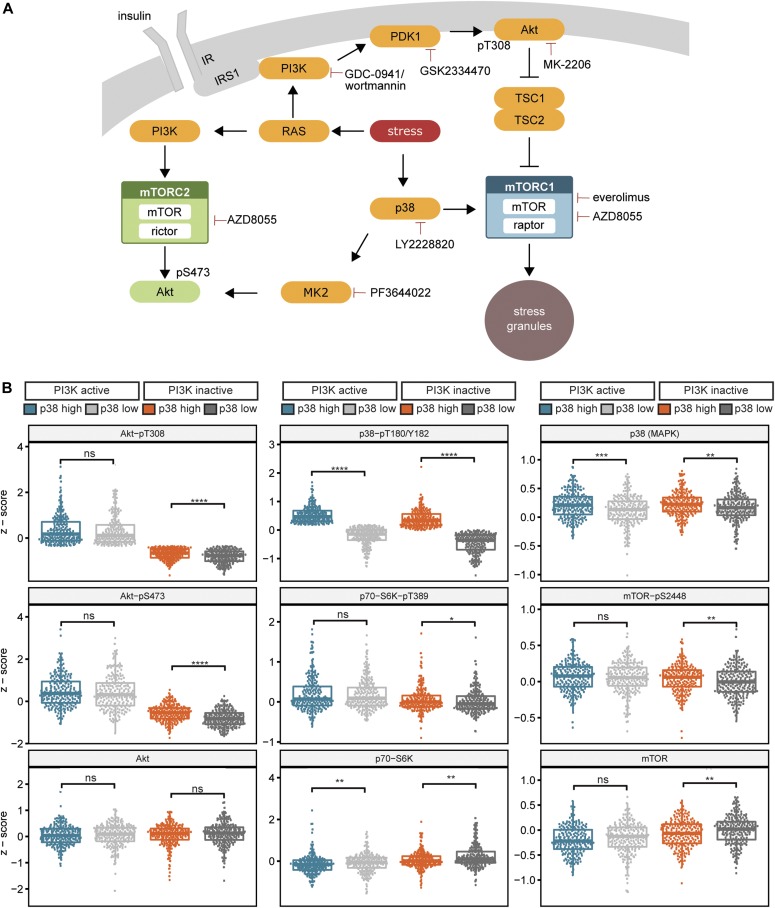

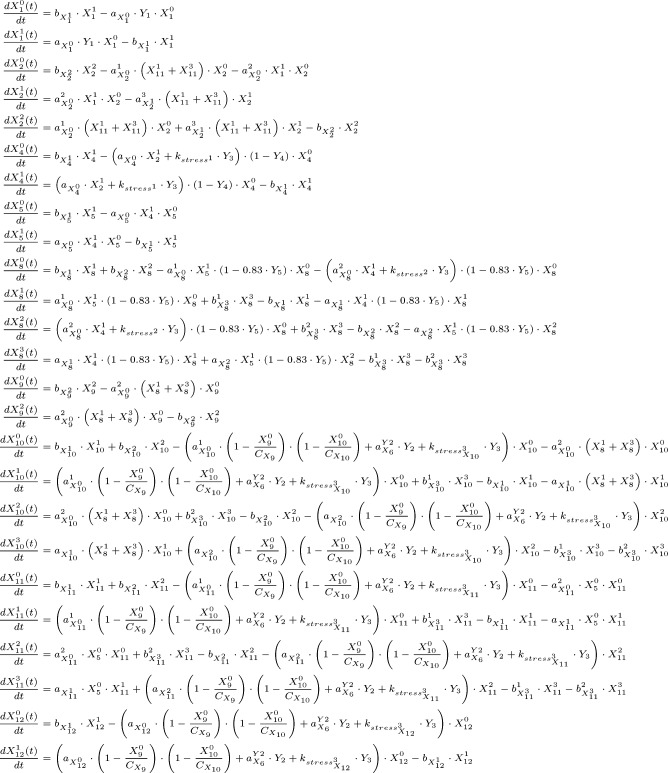
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous