

Dynamics of Gene Expression in Single Root Cells of Arabidopsis thaliana
- PMID: 30923229
- PMCID: PMC8516002
- DOI: 10.1105/tpc.18.00785
Dynamics of Gene Expression in Single Root Cells of Arabidopsis thaliana
Abstract
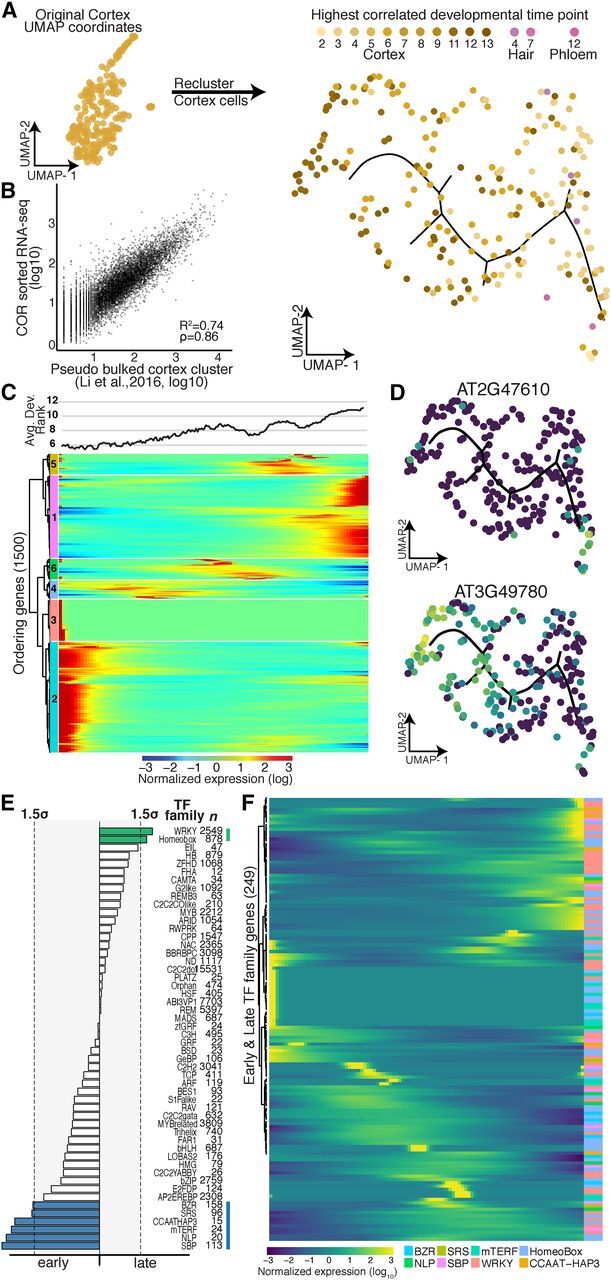
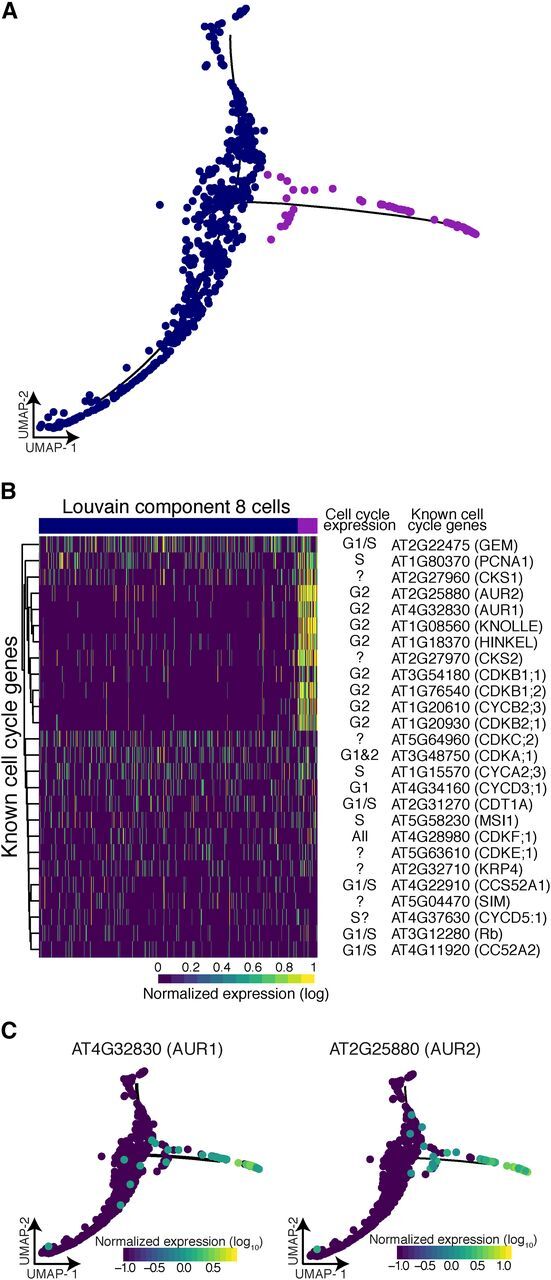
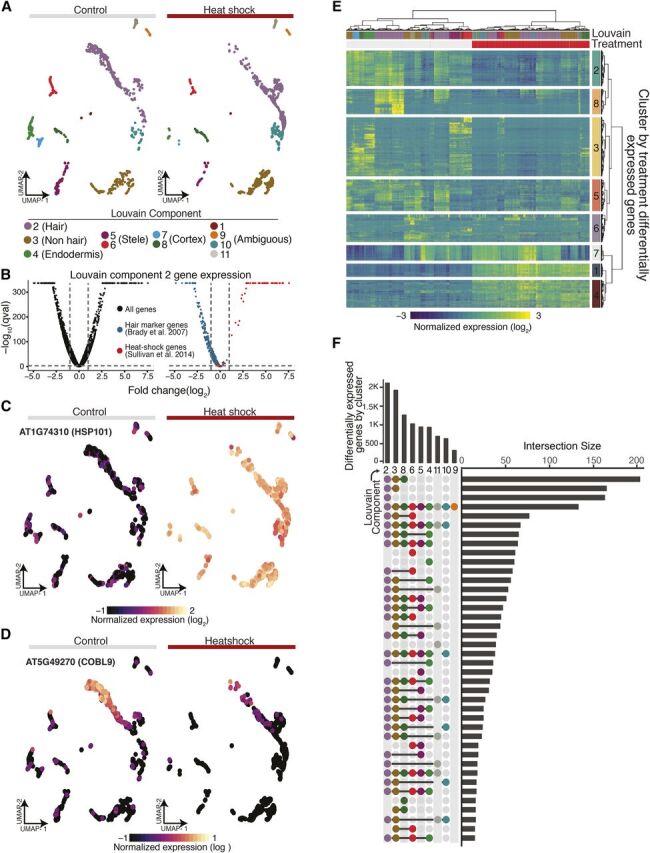
Single cell RNA sequencing can yield high-resolution cell-type-specific expression signatures that reveal new cell types and the developmental trajectories of cell lineages. Here, we apply this approach to Arabidopsis (Arabidopsis thaliana) root cells to capture gene expression in 3,121 root cells. We analyze these data with Monocle 3, which orders single cell transcriptomes in an unsupervised manner and uses machine learning to reconstruct single cell developmental trajectories along pseudotime. We identify hundreds of genes with cell-type-specific expression, with pseudotime analysis of several cell lineages revealing both known and novel genes that are expressed along a developmental trajectory. We identify transcription factor motifs that are enriched in early and late cells, together with the corresponding candidate transcription factors that likely drive the observed expression patterns. We assess and interpret changes in total RNA expression along developmental trajectories and show that trajectory branch points mark developmental decisions. Finally, by applying heat stress to whole seedlings, we address the longstanding question of possible heterogeneity among cell types in the response to an abiotic stress. Although the response of canonical heat-shock genes dominates expression across cell types, subtle but significant differences in other genes can be detected among cell types. Taken together, our results demonstrate that single cell transcriptomics holds promise for studying plant development and plant physiology with unprecedented resolution.
© 2019 All rights reserved.
Figures
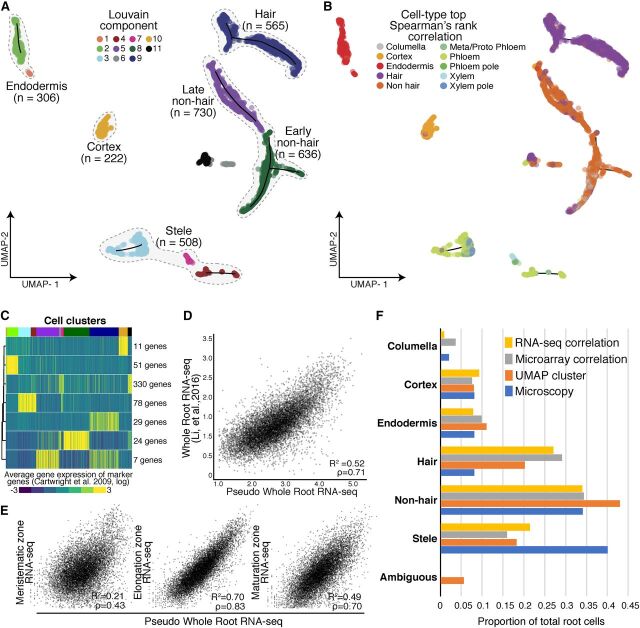
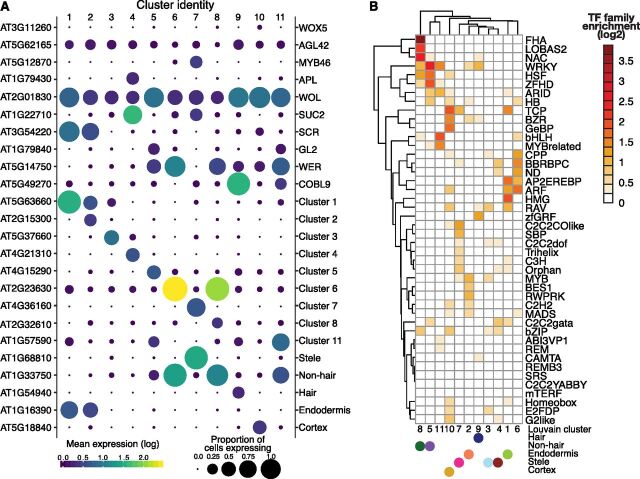
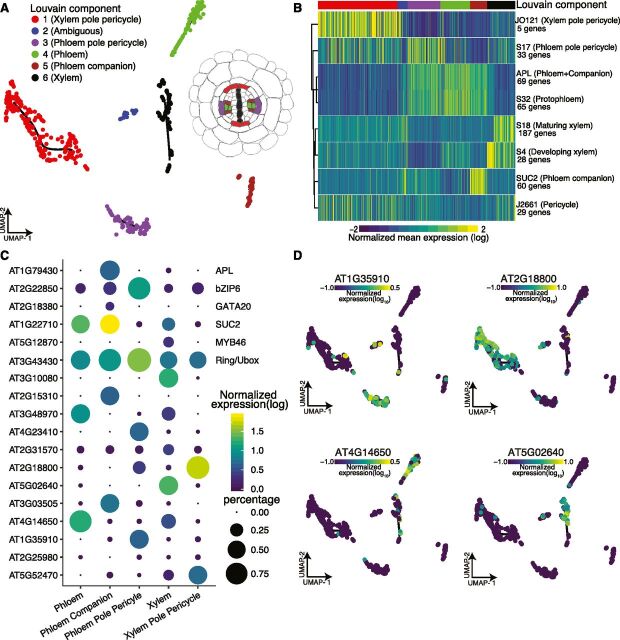
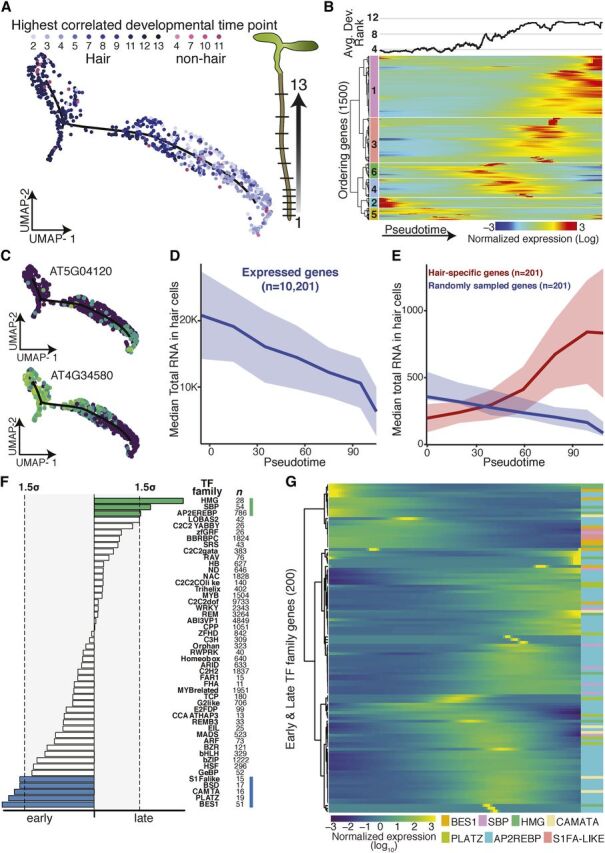

Comment in
-
The Future is Now: Gene Expression Dynamics at Single Cell Resolution.Plant Cell. 2019 May;31(5):933-934. doi: 10.1105/tpc.19.00247. Epub 2019 Apr 12. Plant Cell. 2019. PMID: 30979794 Free PMC article. No abstract available.
References
-
- Birnbaum, K. , Shasha, D.E. , Wang, J.Y. , Jung, J.W. , Lambert, G.M. , Galbraith, D.W. , Benfey, P.N. (2003). A gene expression map of the Arabidopsis root. Science 302: 1956–1960. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
