

Microsatellite Diversity, Complexity, and Host Range of Mycobacteriophage Genomes of the Siphoviridae Family
- PMID: 30923537
- PMCID: PMC6426759
- DOI: 10.3389/fgene.2019.00207
Microsatellite Diversity, Complexity, and Host Range of Mycobacteriophage Genomes of the Siphoviridae Family
Abstract
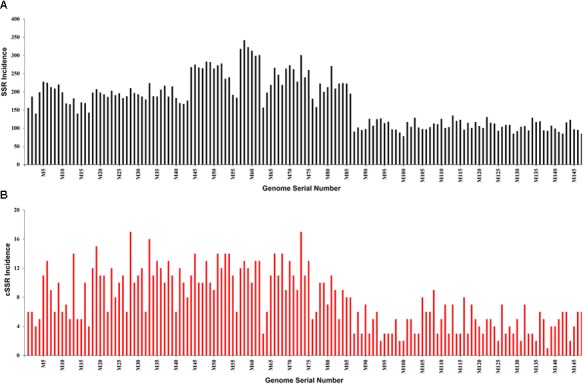
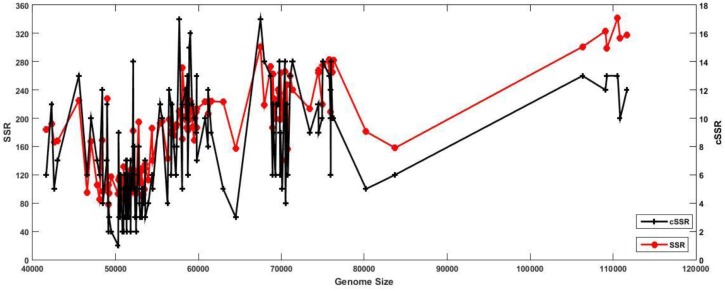
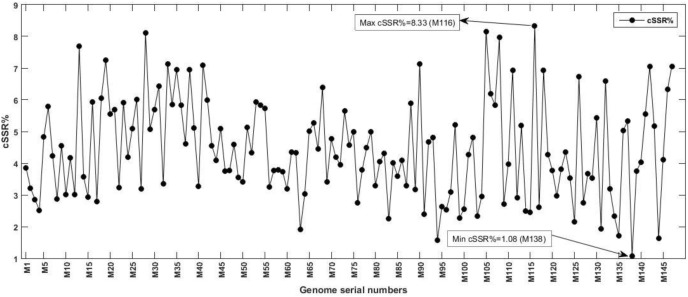
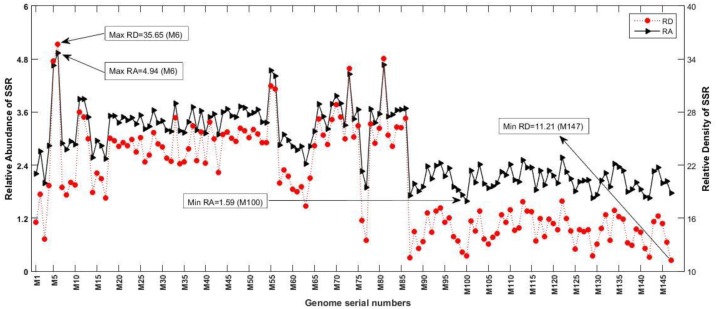
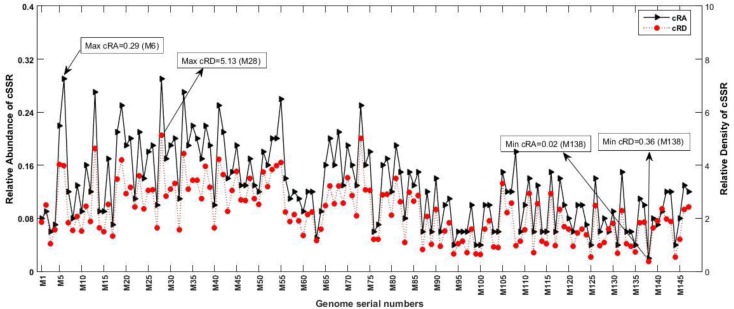
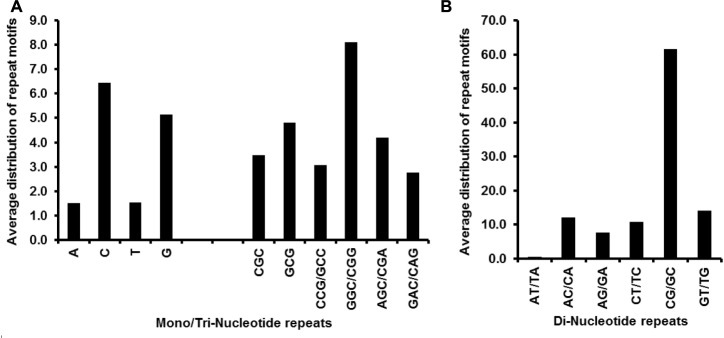
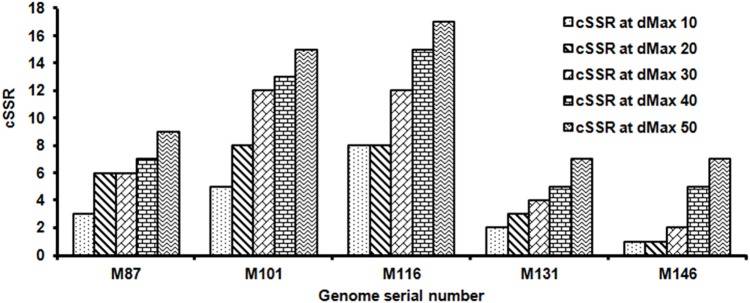
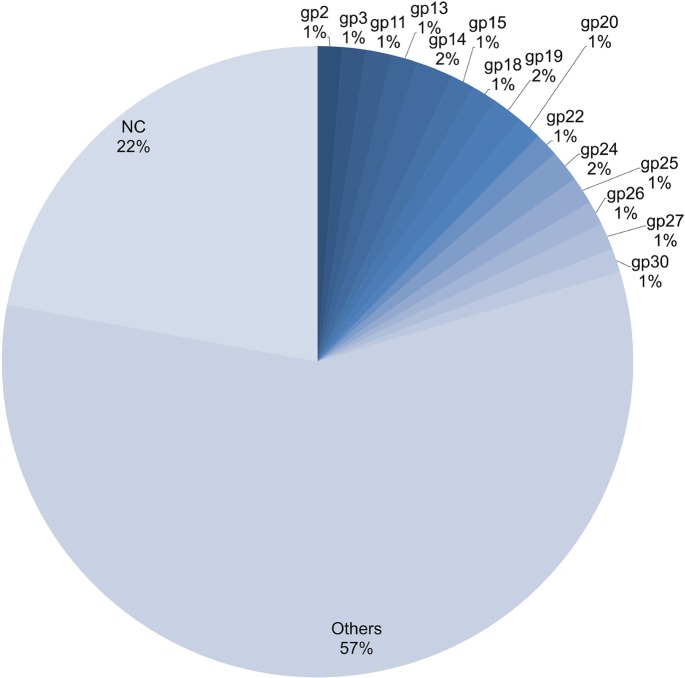
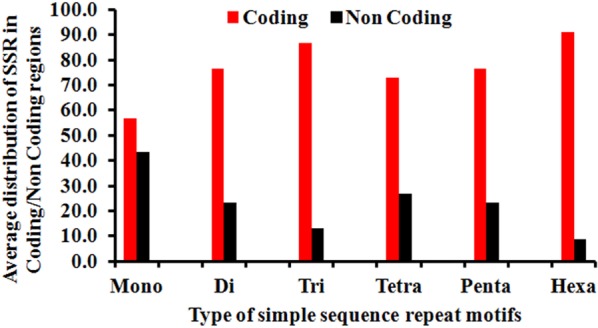
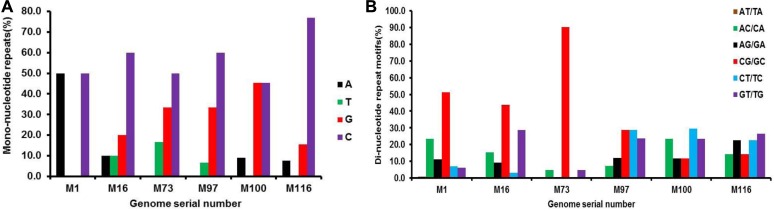
The incidence, distribution, and variation of simple sequence repeats (SSRs) in viruses is instrumental in understanding the functional and evolutionary aspects of repeat sequences. Full-length genome sequences retrieved from NCBI were used for extraction and analysis of repeat sequences using IMEx software. We have also developed two MATLAB-based tools for extraction of gene locations from GenBank in tabular format and simulation of this data with SSR incidence data. Present study encompassing 147 Mycobacteriophage genomes revealed 25,284 SSRs and 1,127 compound SSRs (cSSRs) through IMEx. Mono- to hexa-nucleotide motifs were present. The SSR count per genome ranged from 78 (M100) to 342 (M58) while cSSRs incidence ranged from 1 (M138) to 17 (M28, M73). Though cSSRs were present in all the genomes, their frequency and SSR to cSSR conversion percentage varied from 1.08 (M138 with 93 SSRs) to 8.33 (M116 with 96 SSRs). In terms of localization, the SSRs were predominantly localized to coding regions (∼78%). Interestingly, genomes of around 50 kb contained a similar number of SSRs/cSSRs to that in a 110 kb genome, suggesting functional relevance for SSRs which was substantiated by variation in motif constitution between species with different host range. The three species with broad host range (M97, M100, M116) have around 90% of their mono-nucleotide repeat motifs composed of G or C and only M16 has both A and T mononucleotide motifs. Around 20% of the di-nucleotide repeat motifs in the genomes exhibiting a broad host range were CT/TC, which were either absent or represented to a much lesser extent in the other genomes.
Keywords: Mycobacteriophage; dMAX; host range; imperfect microsatellite extractor; simple sequence repeats.
Figures

References
LinkOut - more resources
Full Text Sources