

Cell lineage and communication network inference via optimization for single-cell transcriptomics
- PMID: 30923815
- PMCID: PMC6582411
- DOI: 10.1093/nar/gkz204
Cell lineage and communication network inference via optimization for single-cell transcriptomics
Abstract
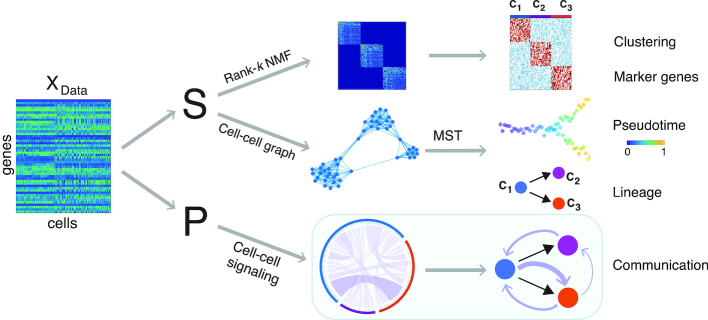
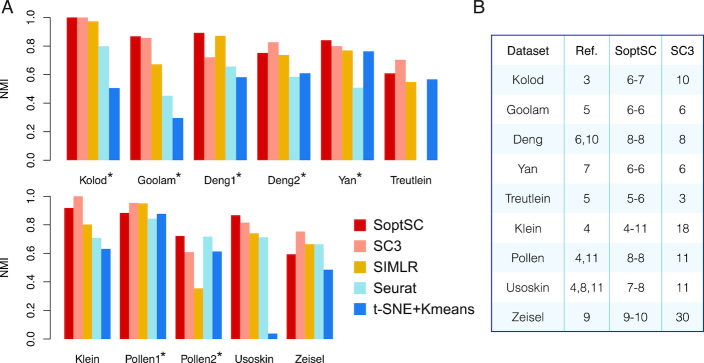
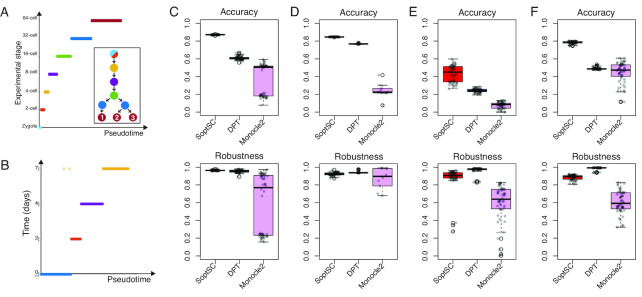
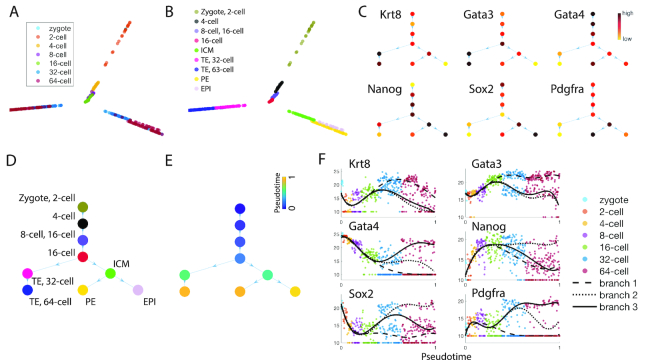
The use of single-cell transcriptomics has become a major approach to delineate cell subpopulations and the transitions between them. While various computational tools using different mathematical methods have been developed to infer clusters, marker genes, and cell lineage, none yet integrate these within a mathematical framework to perform multiple tasks coherently. Such coherence is critical for the inference of cell-cell communication, a major remaining challenge. Here, we present similarity matrix-based optimization for single-cell data analysis (SoptSC), in which unsupervised clustering, pseudotemporal ordering, lineage inference, and marker gene identification are inferred via a structured cell-to-cell similarity matrix. SoptSC then predicts cell-cell communication networks, enabling reconstruction of complex cell lineages that include feedback or feedforward interactions. Application of SoptSC to early embryonic development, epidermal regeneration, and hematopoiesis demonstrates robust identification of subpopulations, lineage relationships, and pseudotime, and prediction of pathway-specific cell communication patterns regulating processes of development and differentiation.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
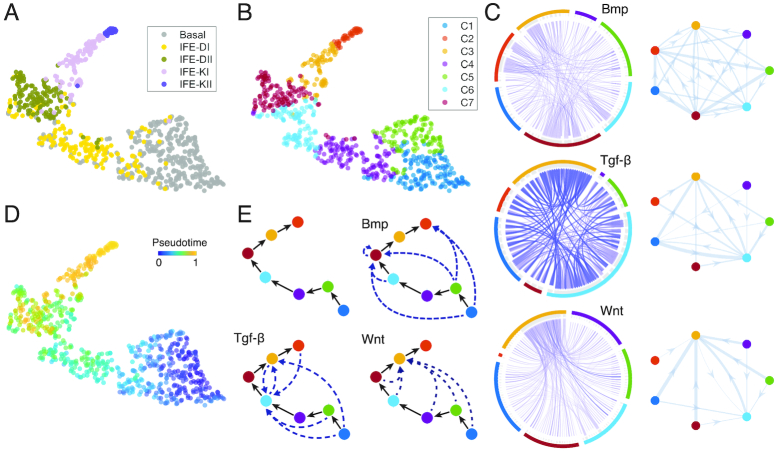
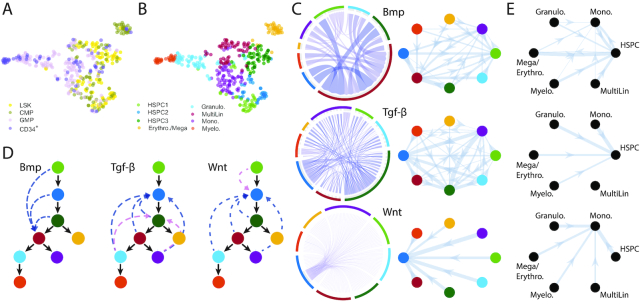
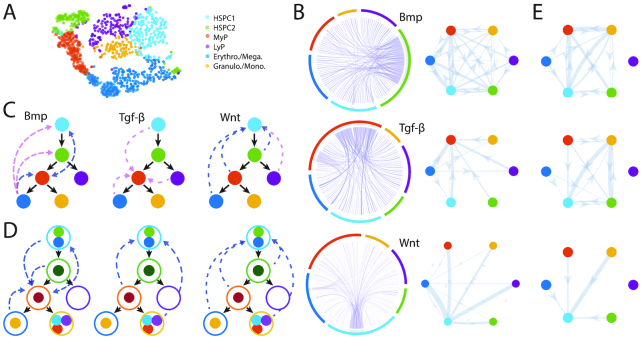
References
-
- Moris N., Pina C., Martinez Arias A.. Transition states and cell fate decisions in epigenetic landscapes. Nat. Rev. Genet. 2016; 17:693–703. - PubMed
-
- Svensson V., Vento-Tormo R., Teichmann S.A.. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018; 13:599–604. - PubMed
-
- Angerer P., Simon L., Tritschler S., Wolf F.A., Fischer D., Theis F.J.. Single cells make big data: New challenges and opportunities in transcriptomics. Curr. Opin. Syst. Biol. 2017; 4:85–91.
