

CYP2C19 and 3A4 Dominate Metabolic Clearance and Bioactivation of Terbinafine Based on Computational and Experimental Approaches
- PMID: 30925039
- PMCID: PMC6692898
- DOI: 10.1021/acs.chemrestox.9b00006
CYP2C19 and 3A4 Dominate Metabolic Clearance and Bioactivation of Terbinafine Based on Computational and Experimental Approaches
Abstract
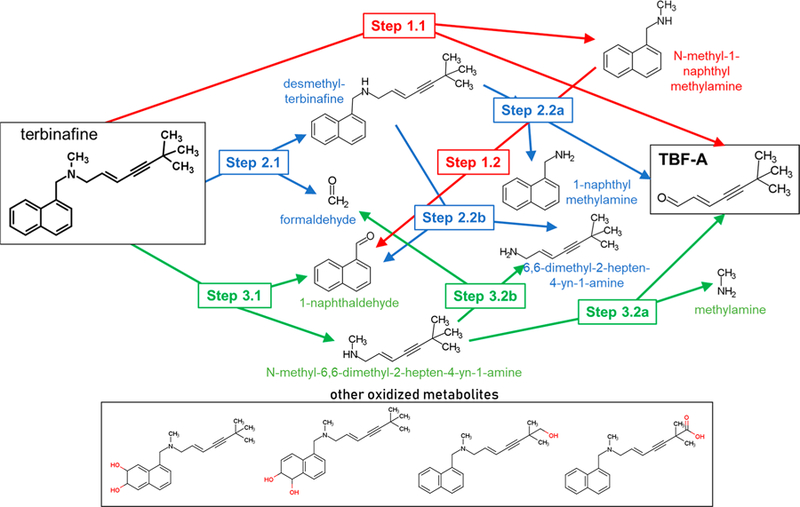
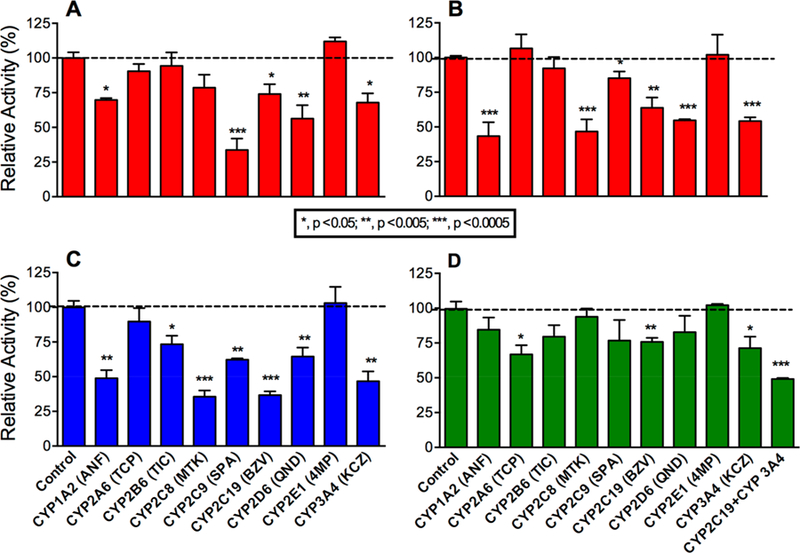
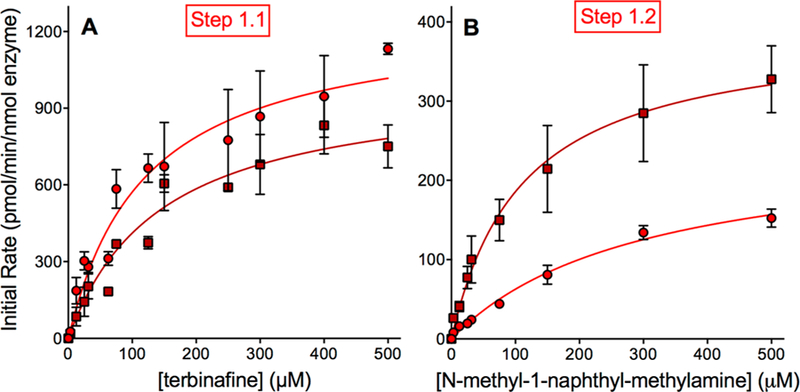
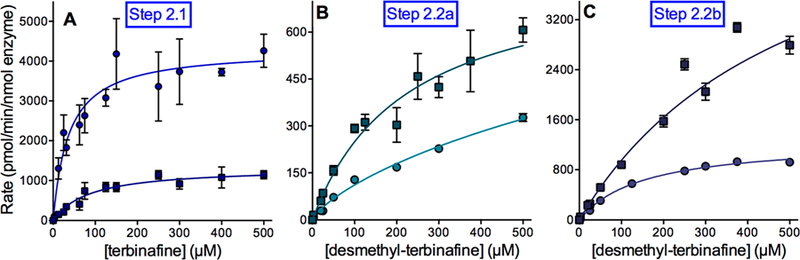
Lamisil (terbinafine) is an effective, widely prescribed antifungal drug that causes rare idiosyncratic hepatotoxicity. The proposed toxic mechanism involves a reactive metabolite, 6,6-dimethyl-2-hepten-4-ynal (TBF-A), formed through three N-dealkylation pathways. We were the first to characterize them using in vitro studies with human liver microsomes and modeling approaches, yet knowledge of the individual enzymes catalyzing reactions remained unknown. Herein, we employed experimental and computational tools to assess terbinafine metabolism by specific cytochrome P450 isozymes. In vitro inhibitor phenotyping studies revealed six isozymes were involved in one or more N-dealkylation pathways. CYP2C19 and 3A4 contributed to all pathways, and so, we targeted them for steady-state analyses with recombinant isozymes. N-Dealkylation yielding TBF-A directly was catalyzed by CYP2C19 and 3A4 similarly. Nevertheless, CYP2C19 was more efficient than CYP3A4 at N-demethylation and other steps leading to TBF-A. Unlike microsomal reactions, N-denaphthylation was surprisingly efficient for CYP2C19 and 3A4, which was validated by controls. CYP2C19 was the most efficient among all reactions. Nonetheless, CYP3A4 was more selective at steps leading to TBF-A, making it more effective in terbinafine bioactivation based on metabolic split ratios for competing pathways. Model predictions did not extrapolate to quantitative kinetic constants, yet some results for CYP3A4 and CYP2C19 agreed qualitatively with preferred reaction steps and pathways. Clinical data on drug interactions support the CYP3A4 role in terbinafine metabolism, while CYP2C19 remains understudied. Taken together, knowledge of P450s responsible for terbinafine metabolism and TBF-A formation provides a foundation for investigating and mitigating the impact of P450 variations in toxic risks posed to patients.
Conflict of interest statement
Notes
The authors declare no competing financial interest.
Figures
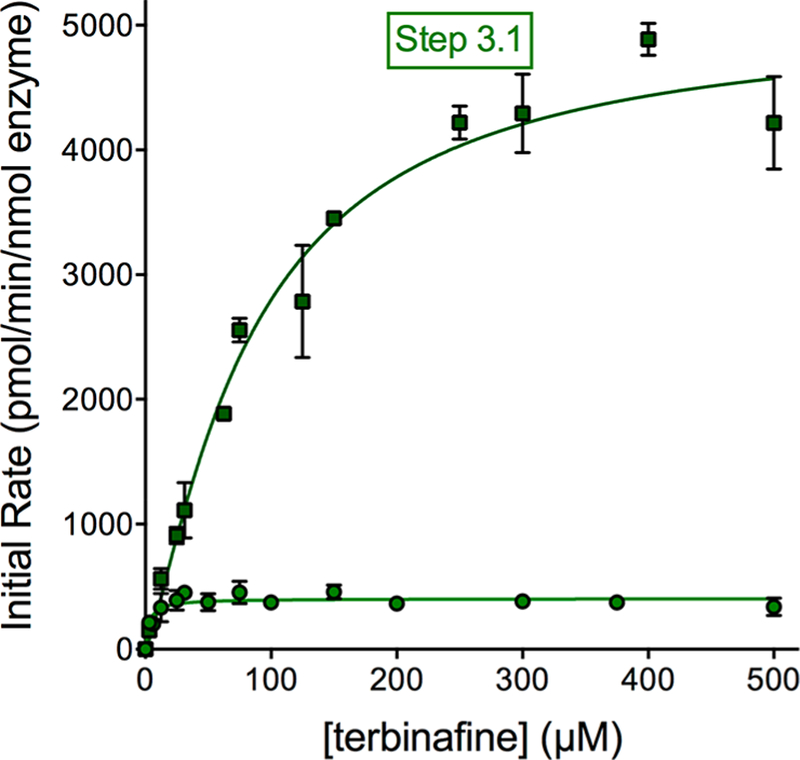
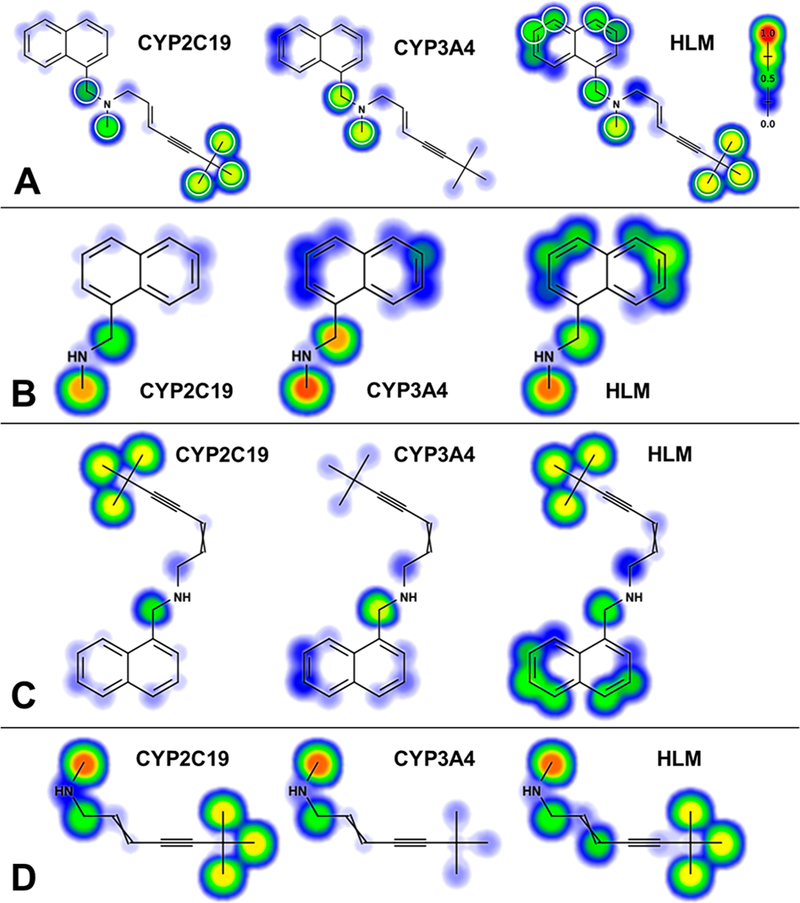
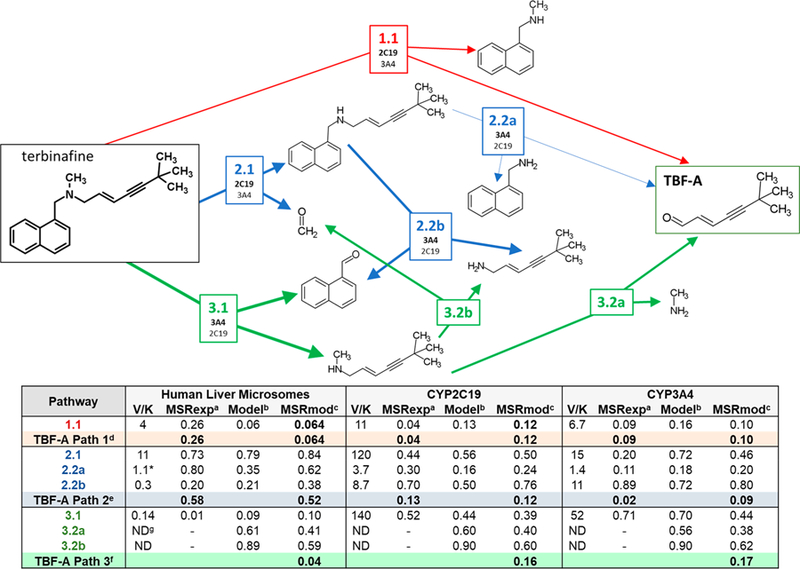
References
-
- Ryder NS (1992) Terbinafine: Mode of Action and Properties of the Squalene Epoxidase Inhibition. Br. J. Dermatol 126 (s39), 2–7. - PubMed
-
- Nowosielski M, Hoffmann M, Wyrwicz LS, Stepniak P, Plewczynski DM, Lazniewski M, Ginalski K, and Rychlewski L (2011) Detailed Mechanism of Squalene Epoxidase Inhibition by Terbinafine. J. Chem. Inf. Model 51 (2), 455–462. - PubMed
-
- Hall M, Monka C, Krupp P, and O’Sullivan D (1997) Safety of Oral Terbinafine: Results of a Postmarketing Surveillance Study in 25 884 Patients. Arch. Dermatol 133 (10), 1213–1219. - PubMed
-
- Gupta AK, Sibbald RG, Knowles SR, Lynde CW, and Shear NH (1997) Terbinafine Therapy May Be Associated with the Development of Psoriasis de Novo or Its Exacerbation: Four Case Reports and a Review of Drug-Induced Psoriasis. J. Am. Acad. Dermatol 36 (5), 858–862. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
