

Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis
- PMID: 30926780
- PMCID: PMC6441045
- DOI: 10.1038/s41467-019-09139-4
Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis
Abstract
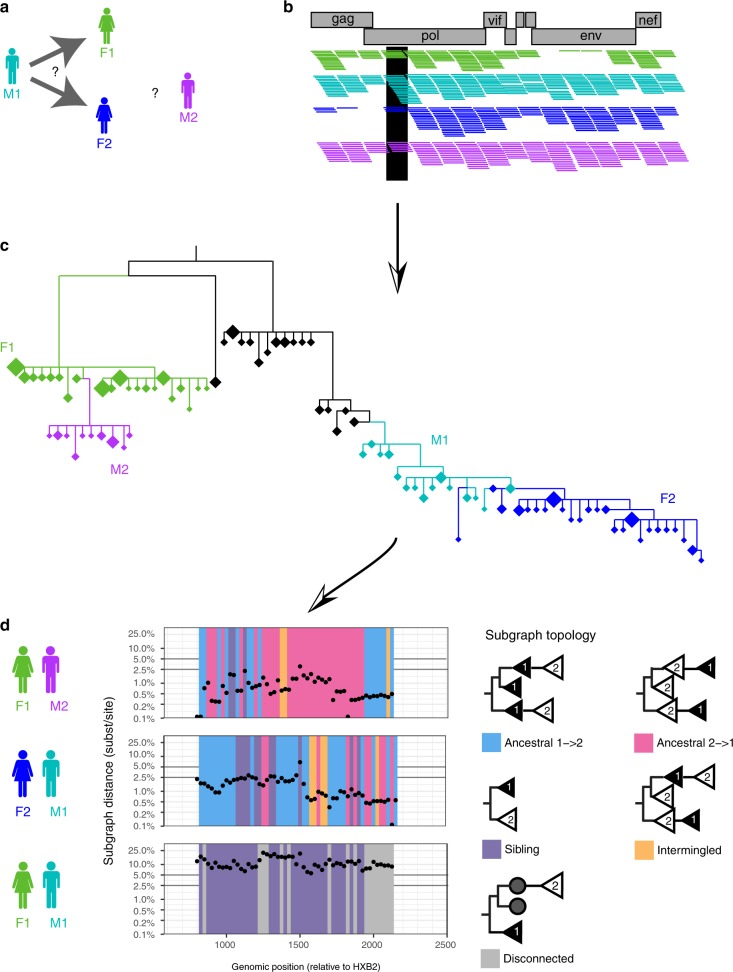
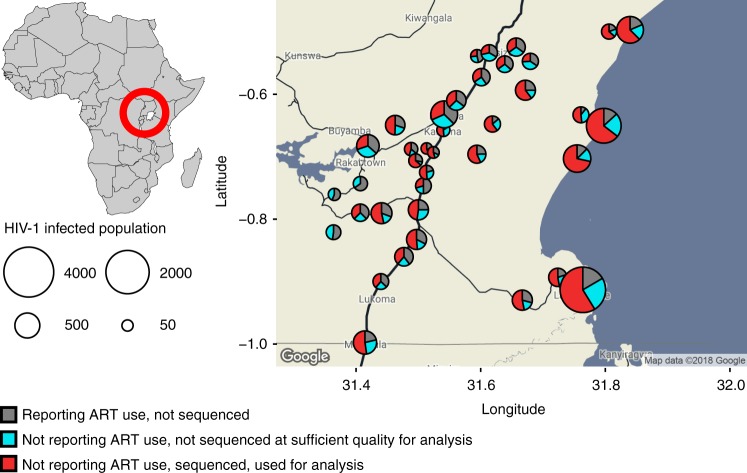
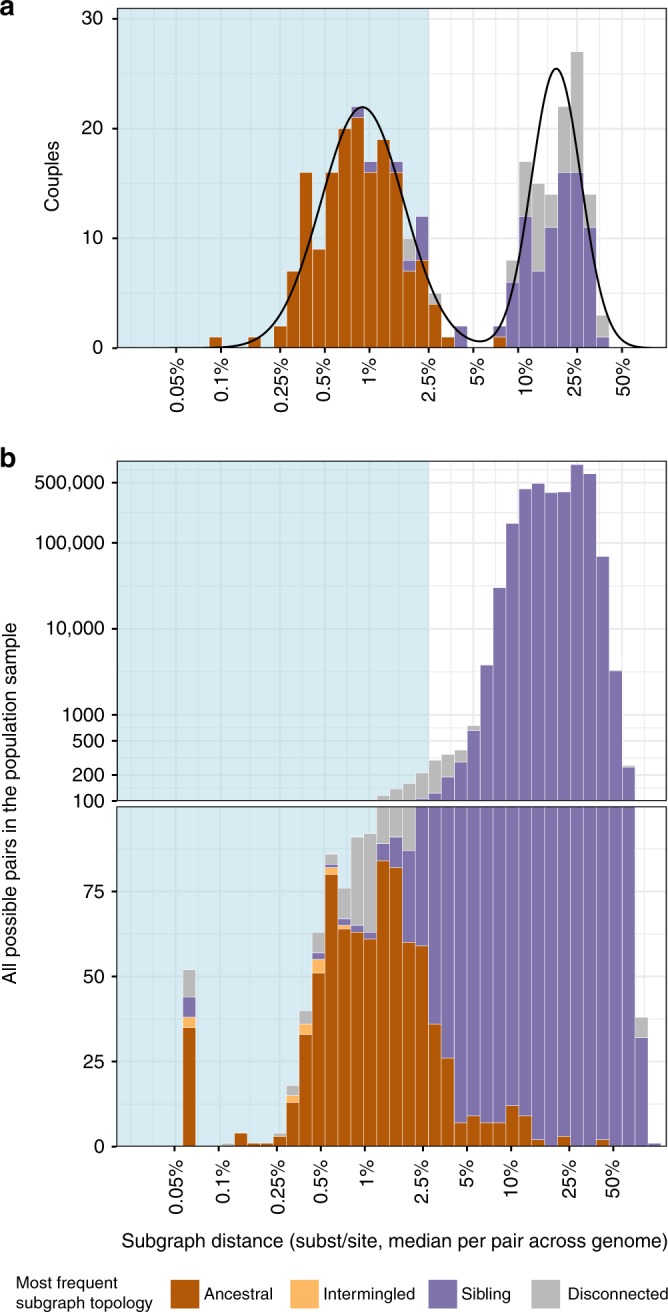
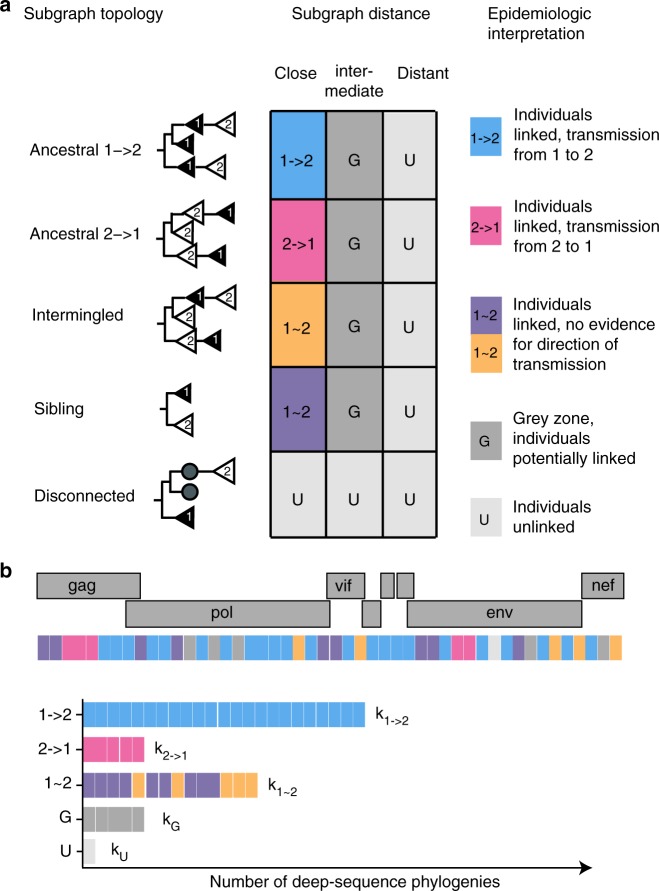
To prevent new infections with human immunodeficiency virus type 1 (HIV-1) in sub-Saharan Africa, UNAIDS recommends targeting interventions to populations that are at high risk of acquiring and passing on the virus. Yet it is often unclear who and where these 'source' populations are. Here we demonstrate how viral deep-sequencing can be used to reconstruct HIV-1 transmission networks and to infer the direction of transmission in these networks. We are able to deep-sequence virus from a large population-based sample of infected individuals in Rakai District, Uganda, reconstruct partial transmission networks, and infer the direction of transmission within them at an estimated error rate of 16.3% [8.8-28.3%]. With this error rate, deep-sequence phylogenetics cannot be used against individuals in legal contexts, but is sufficiently low for population-level inferences into the sources of epidemic spread. The technique presents new opportunities for characterizing source populations and for targeting of HIV-1 prevention interventions in Africa.
Conflict of interest statement
The authors declare no competing interests.
Figures
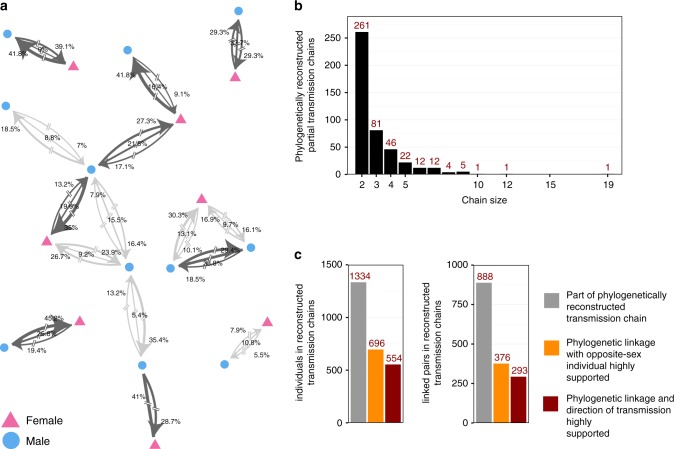
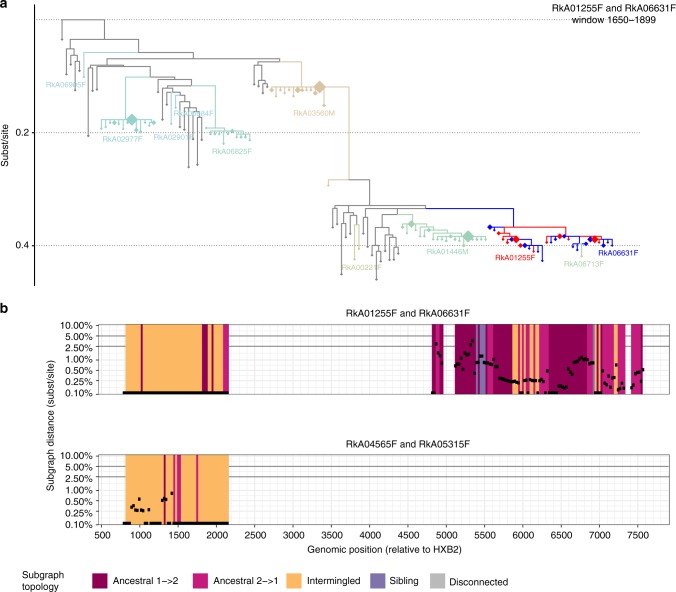
References
-
- UNAIDS. UNAIDS Data 2017, Document JC2910E. http://www.unaids.org/en/resources/documents/2017/2017_data_book (2017).
-
- UNAIDS. Fast-track: ending the AIDS epidemic by 2030, Document JC2686. http://www.unaids.org/en/resources/documents/2014/JC2686_WAD2014report (2014).
-
- UNAIDS. Empower young women and adolescent girls: fast-track the end of the AIDS epidemic in Africa, Document JC2746. http://www.unaids.org/en/resources/documents/2015/JC2746 (2015).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
