

Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions
- PMID: 30929737
- PMCID: PMC6451739
- DOI: 10.1016/j.ajhg.2019.03.008
Diagnostic Utility of Genome-wide DNA Methylation Testing in Genetically Unsolved Individuals with Suspected Hereditary Conditions
Abstract
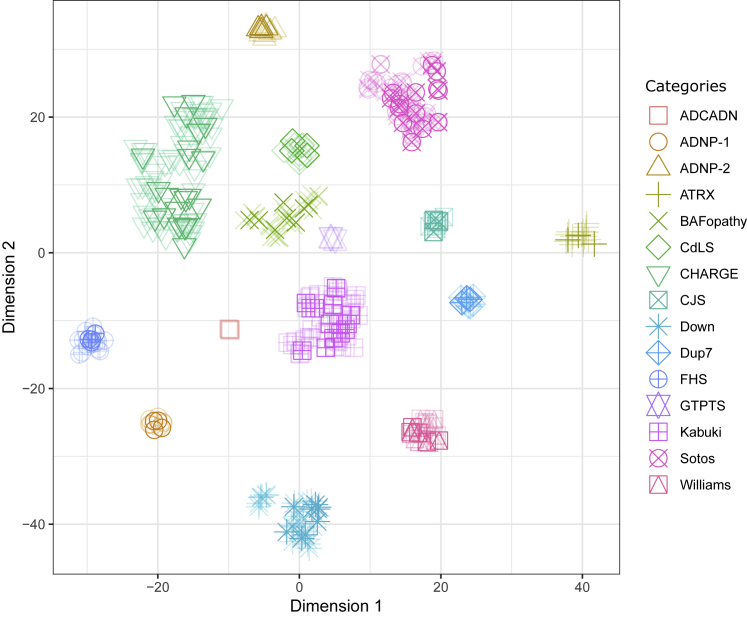
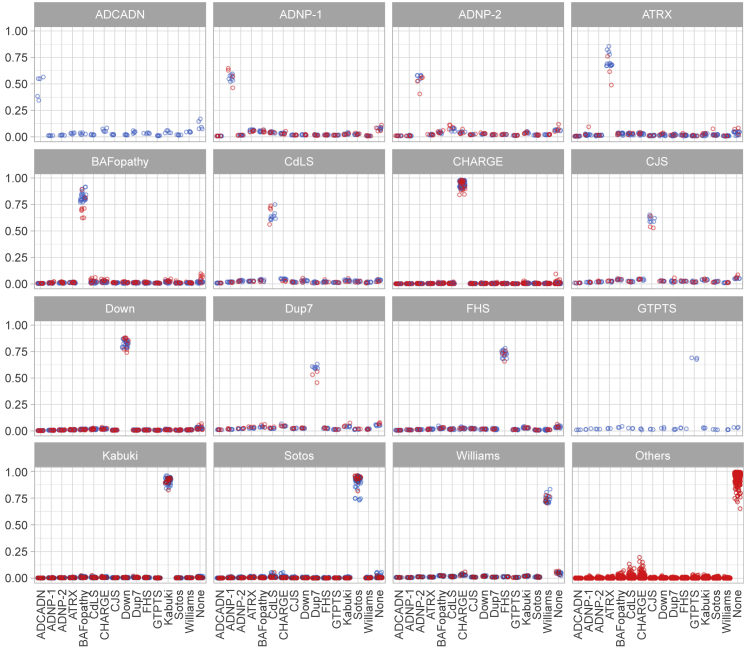
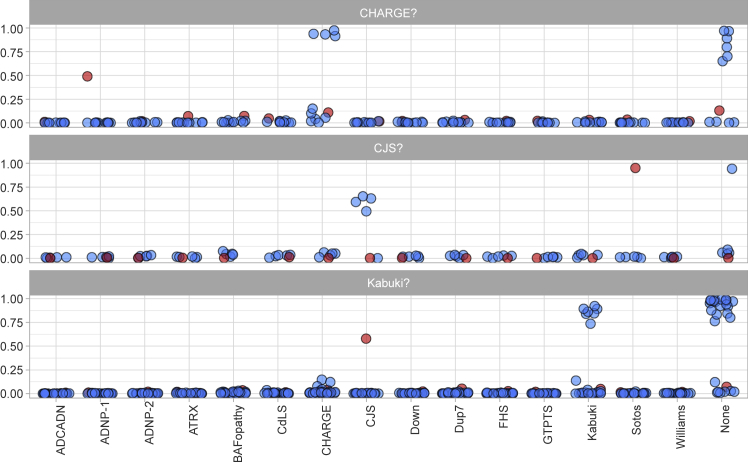
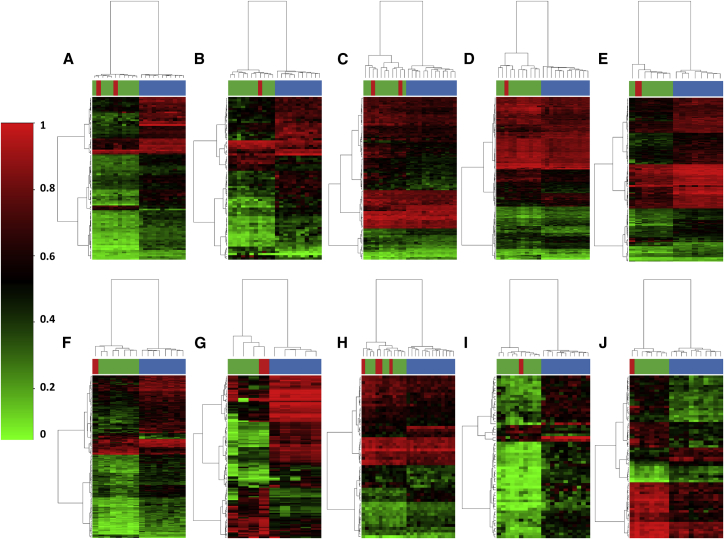
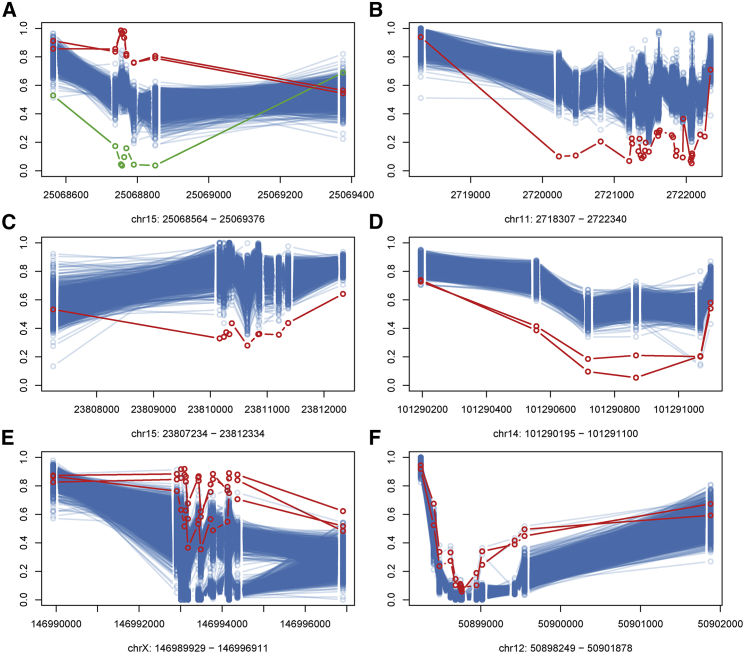
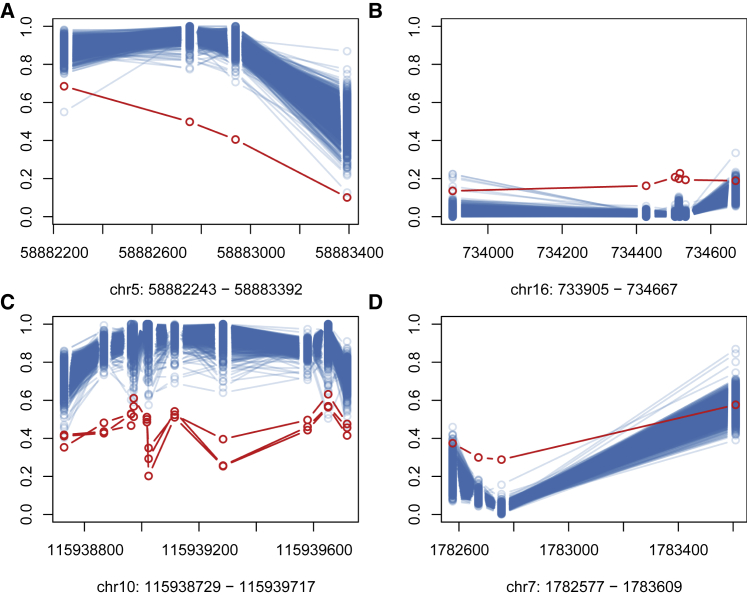
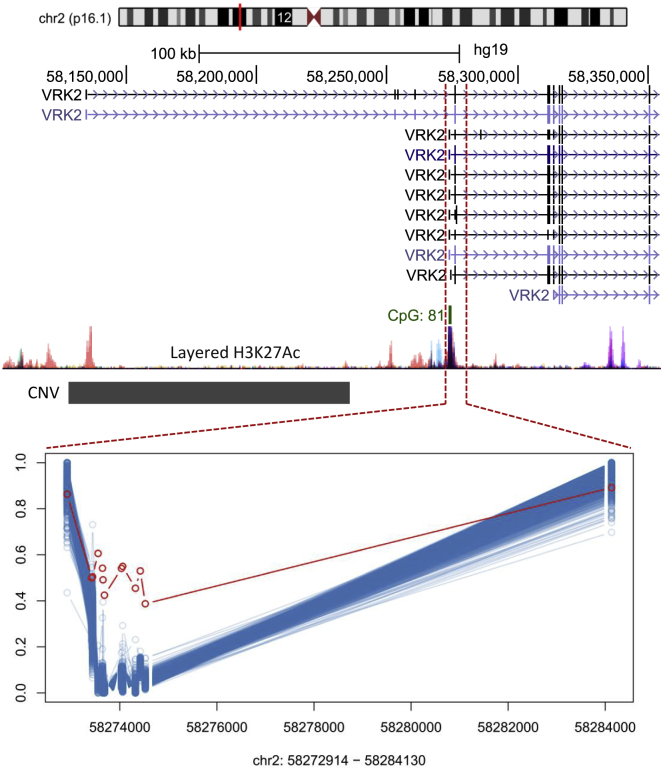
Conventional genetic testing of individuals with neurodevelopmental presentations and congenital anomalies (ND/CAs), i.e., the analysis of sequence and copy number variants, leaves a substantial proportion of them unexplained. Some of these cases have been shown to result from DNA methylation defects at a single locus (epi-variants), while others can exhibit syndrome-specific DNA methylation changes across multiple loci (epi-signatures). Here, we investigate the clinical diagnostic utility of genome-wide DNA methylation analysis of peripheral blood in unresolved ND/CAs. We generate a computational model enabling concurrent detection of 14 syndromes using DNA methylation data with full accuracy. We demonstrate the ability of this model in resolving 67 individuals with uncertain clinical diagnoses, some of whom had variants of unknown clinical significance (VUS) in the related genes. We show that the provisional diagnoses can be ruled out in many of the case subjects, some of whom are shown by our model to have other diseases initially not considered. By applying this model to a cohort of 965 ND/CA-affected subjects without a previous diagnostic assumption and a separate assessment of rare epi-variants in this cohort, we identify 15 case subjects with syndromic Mendelian disorders, 12 case subjects with imprinting and trinucleotide repeat expansion disorders, as well as 106 case subjects with rare epi-variants, a portion of which involved genes clinically or functionally linked to the subjects' phenotypes. This study demonstrates that genomic DNA methylation analysis can facilitate the molecular diagnosis of unresolved clinical cases and highlights the potential value of epigenomic testing in the routine clinical assessment of ND/CAs.
Keywords: DNA methylation; clinical epigenomic testing; congenital anomaly; epi-signature; epi-variant; imprinting; multiclass prediction; neurodevelopmental syndrome; nucleotide expansion repeat; unresolved case; variant of unknown significance.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Rodriguez-Revenga L., Mila M., Rosenberg C., Lamb A., Lee C. Structural variation in the human genome: the impact of copy number variants on clinical diagnosis. Genet. Med. 2007;9:600–606. - PubMed
-
- Gilissen C., Hehir-Kwa J.Y., Thung D.T., van de Vorst M., van Bon B.W., Willemsen M.H., Kwint M., Janssen I.M., Hoischen A., Schenck A. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–347. - PubMed
-
- Eisenberger T., Neuhaus C., Khan A.O., Decker C., Preising M.N., Friedburg C., Bieg A., Gliem M., Charbel Issa P., Holz F.G. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PLoS ONE. 2013;8:e78496. - PMC - PubMed
-
- Aref-Eshghi E., Schenkel L.C., Carere D.A., Rodenhiser D.I., Sadikovic B. Second Edition; 2018. Epigenomic Mechanisms of Human Developmental Disorders. In Epigenetics in Human Disease; pp. 837–859.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
