

Truncating Mutations in UBAP1 Cause Hereditary Spastic Paraplegia
- PMID: 30929741
- PMCID: PMC6451742
- DOI: 10.1016/j.ajhg.2019.03.001
Truncating Mutations in UBAP1 Cause Hereditary Spastic Paraplegia
Erratum in
-
Truncating Mutations in UBAP1 Cause Hereditary Spastic Paraplegia.Am J Hum Genet. 2019 Jun 6;104(6):1251. doi: 10.1016/j.ajhg.2019.05.009. Am J Hum Genet. 2019. PMID: 31173719 Free PMC article. No abstract available.
Abstract
The diagnostic gap for rare neurodegenerative diseases is still considerable, despite continuous advances in gene identification. Many novel Mendelian genes have only been identified in a few families worldwide. Here we report the identification of an autosomal-dominant gene for hereditary spastic paraplegia (HSP) in 10 families that are of diverse geographic origin and whose affected members all carry unique truncating changes in a circumscript region of UBAP1 (ubiquitin-associated protein 1). HSP is a neurodegenerative disease characterized by progressive lower-limb spasticity and weakness, as well as frequent bladder dysfunction. At least 40% of affected persons are currently undiagnosed after exome sequencing. We identified pathological truncating variants in UBAP1 in affected persons from Iran, USA, Germany, Canada, Spain, and Bulgarian Roma. The genetic support ranges from linkage in the largest family (LOD = 8.3) to three confirmed de novo mutations. We show that mRNA in the fibroblasts of affected individuals escapes nonsense-mediated decay and thus leads to the expression of truncated proteins; in addition, concentrations of the full-length protein are reduced in comparison to those in controls. This suggests either a dominant-negative effect or haploinsufficiency. UBAP1 links endosomal trafficking to the ubiquitination machinery pathways that have been previously implicated in HSPs, and UBAP1 provides a bridge toward a more unified pathophysiology.
Keywords: animal model; endosomal trafficking; genetic diseases; hereditary spastic paraplegia; neurodegenerative diseases; spasticity; ubiquitination; zebrafish.
Copyright © 2019. Published by Elsevier Inc.
Figures
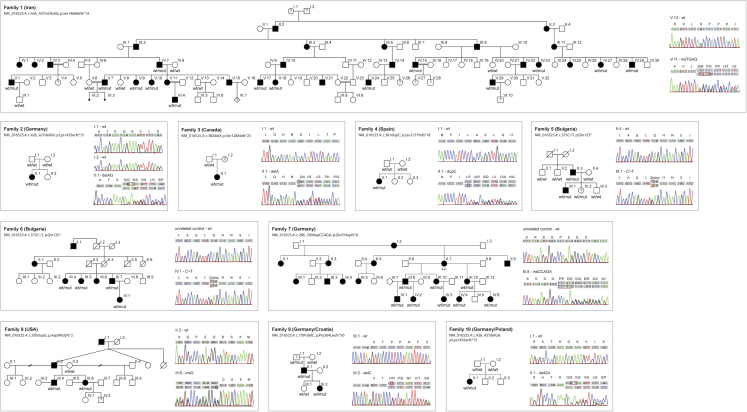
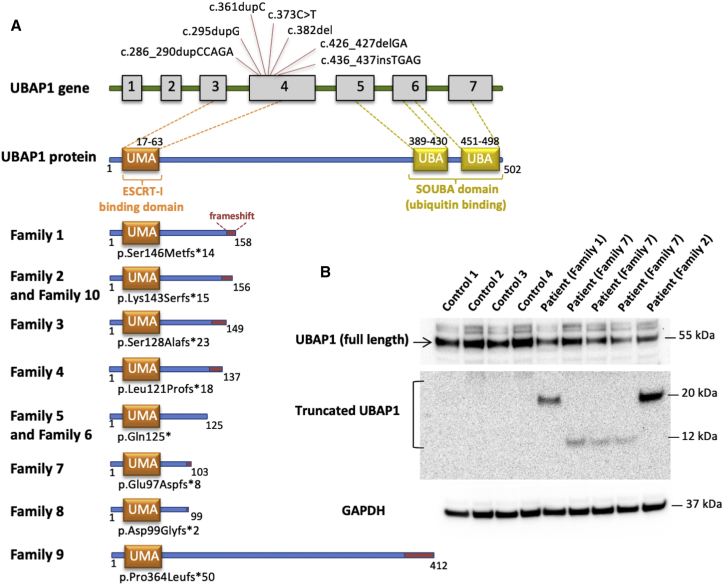
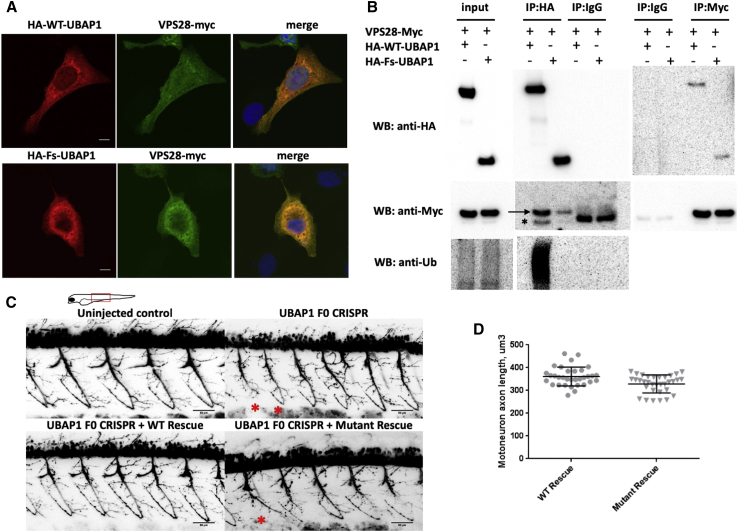
References
-
- Parodi L., Fenu S., Stevanin G., Durr A. Hereditary spastic paraplegia: More than an upper motor neuron disease. Rev. Neurol. (Paris) 2017;173:352–360. - PubMed
-
- Schüle R., Wiethoff S., Martus P., Karle K.N., Otto S., Klebe S., Klimpe S., Gallenmüller C., Kurzwelly D., Henkel D. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann. Neurol. 2016;79:646–658. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
