

Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome
- PMID: 30931400
- PMCID: PMC6436731
- DOI: 10.1042/ETLS20180100
Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome
Abstract
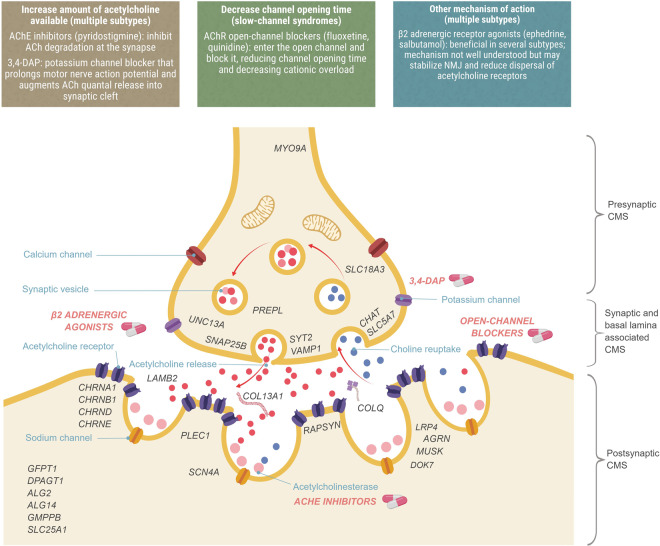
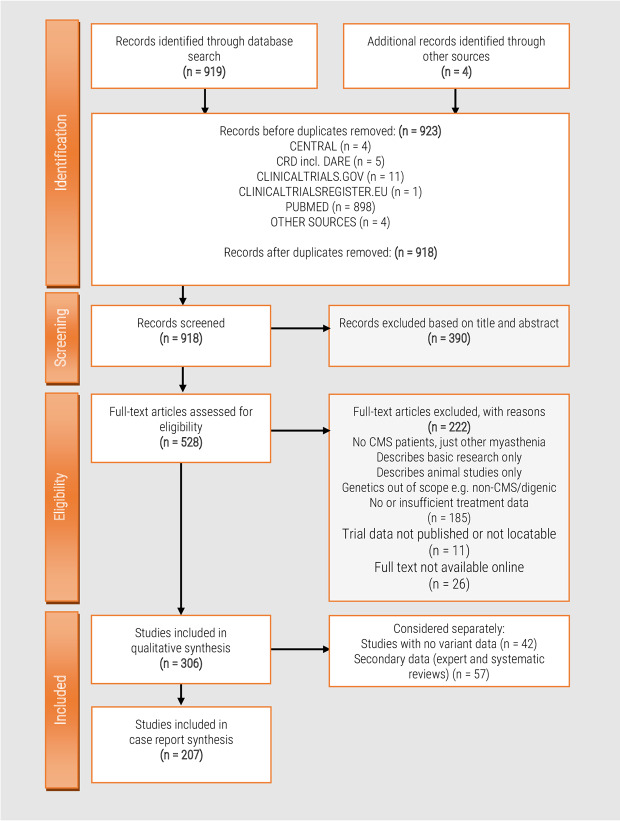
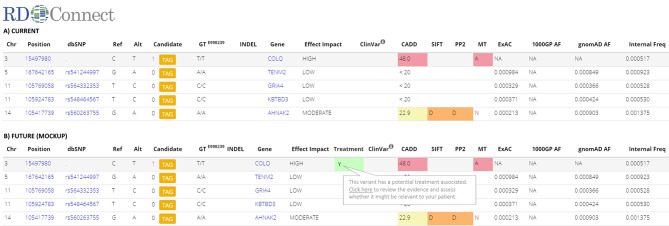
Despite recent scientific advances, most rare genetic diseases - including most neuro-muscular diseases - do not currently have curative gene-based therapies available. However, in some cases, such as vitamin, cofactor or enzyme deficiencies, channelopathies and disorders of the neuromuscular junction, a confirmed genetic diagnosis provides guidance on treatment, with drugs available that may significantly alter the disease course, improve functional ability and extend life expectancy. Nevertheless, many treatable patients remain undiagnosed or do not receive treatment even after genetic diagnosis. The growth of computer-aided genetic analysis systems that enable clinicians to diagnose their undiagnosed patients has not yet been matched by genetics-based decision-support systems for treatment guidance. Generating a 'treatabolome' of treatable variants and the evidence for the treatment has the potential to increase treatment rates for treatable conditions. Here, we use the congenital myasthenic syndromes (CMS), a group of clinically and genetically heterogeneous but frequently treatable neuromuscular conditions, to illustrate the steps in the creation of a treatabolome for rare inherited diseases. We perform a systematic review of the evidence for pharmacological treatment of each CMS type, gathering evidence from 207 studies of over 1000 patients and stratifying by genetic defect, as treatment varies depending on the underlying cause. We assess the strength and quality of the evidence and create a dataset that provides the foundation for a computer-aided system to enable clinicians to gain easier access to information about treatable variants and the evidence they need to consider.
Conflict of interest statement
Competing Interests The Authors declare that there are no competing interests associated with the manuscript.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources