

A small-molecule inhibitor of the DNA recombinase Rad51 from Plasmodium falciparum synergizes with the antimalarial drugs artemisinin and chloroquine
- PMID: 30936202
- PMCID: PMC6527153
- DOI: 10.1074/jbc.RA118.005009
A small-molecule inhibitor of the DNA recombinase Rad51 from Plasmodium falciparum synergizes with the antimalarial drugs artemisinin and chloroquine
Abstract
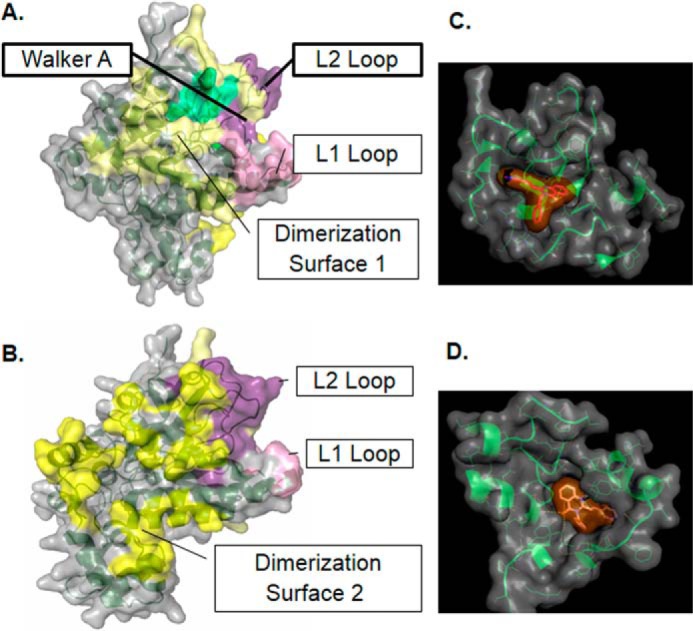
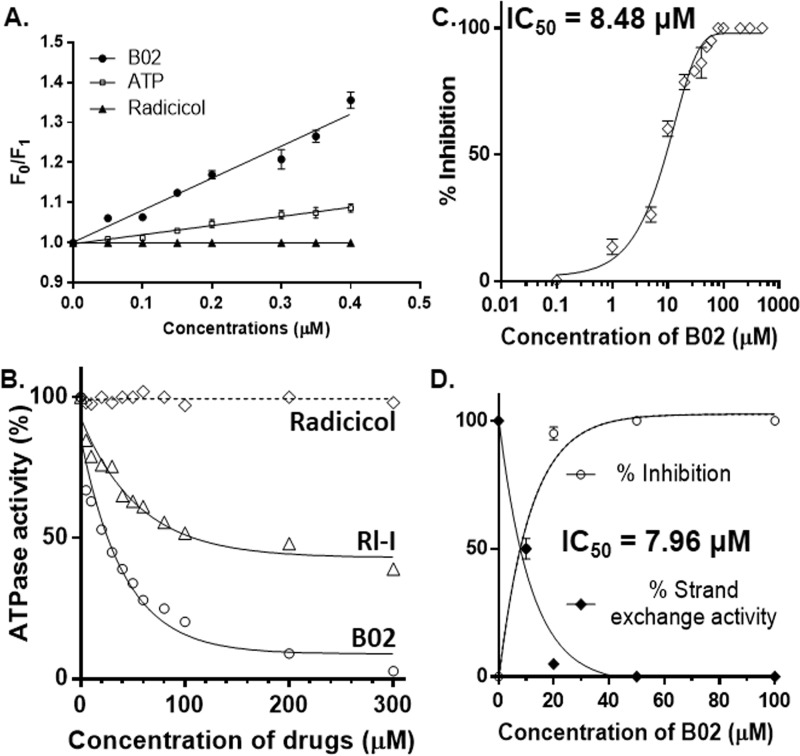
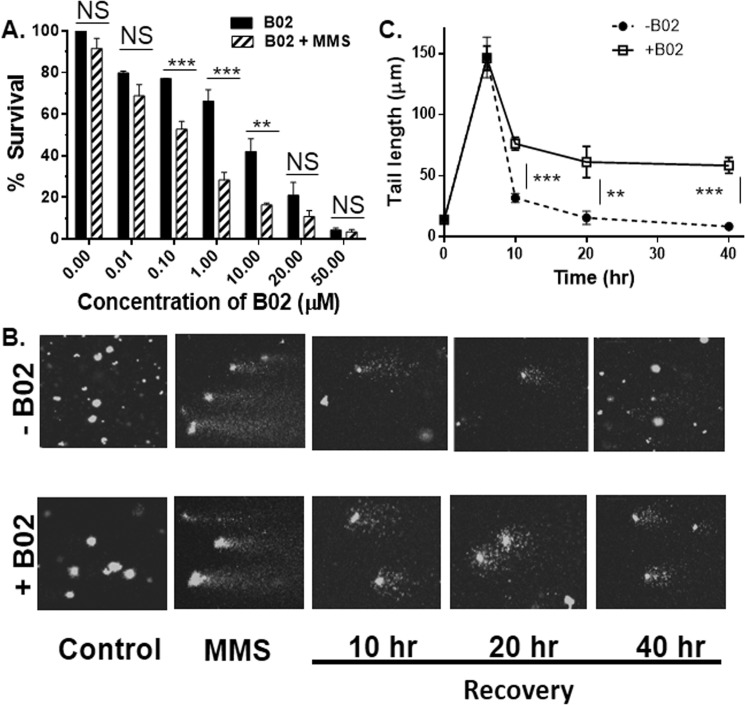
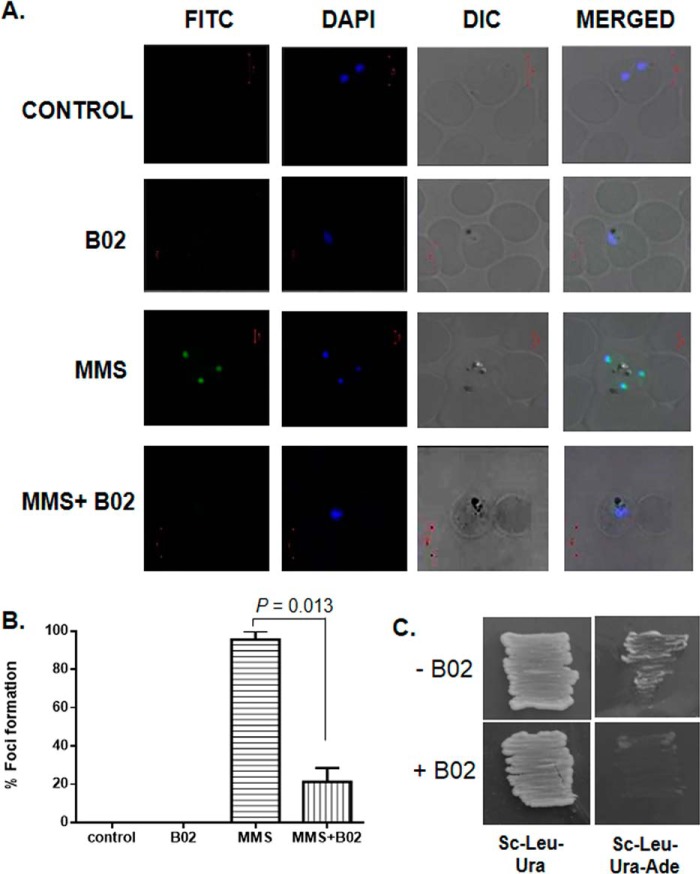
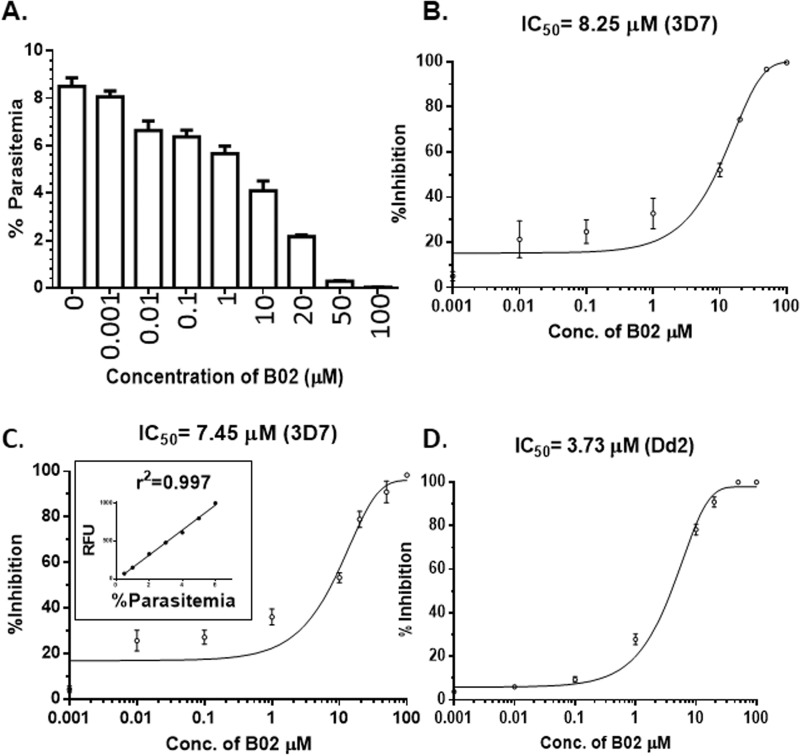
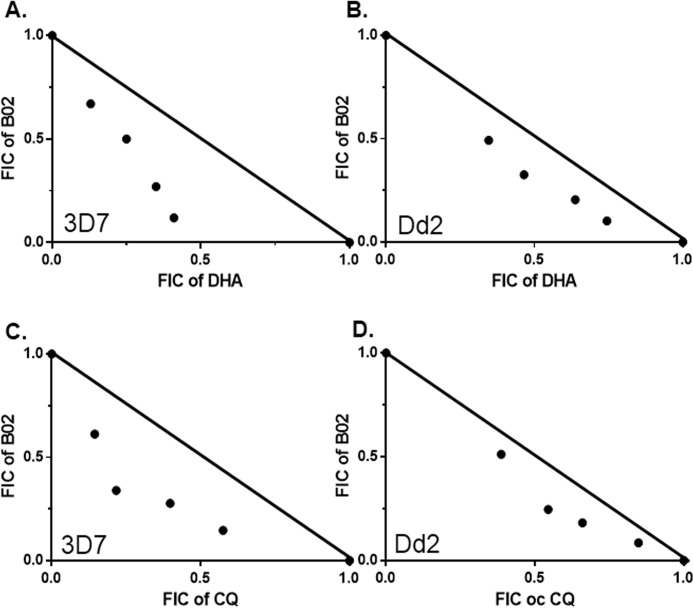
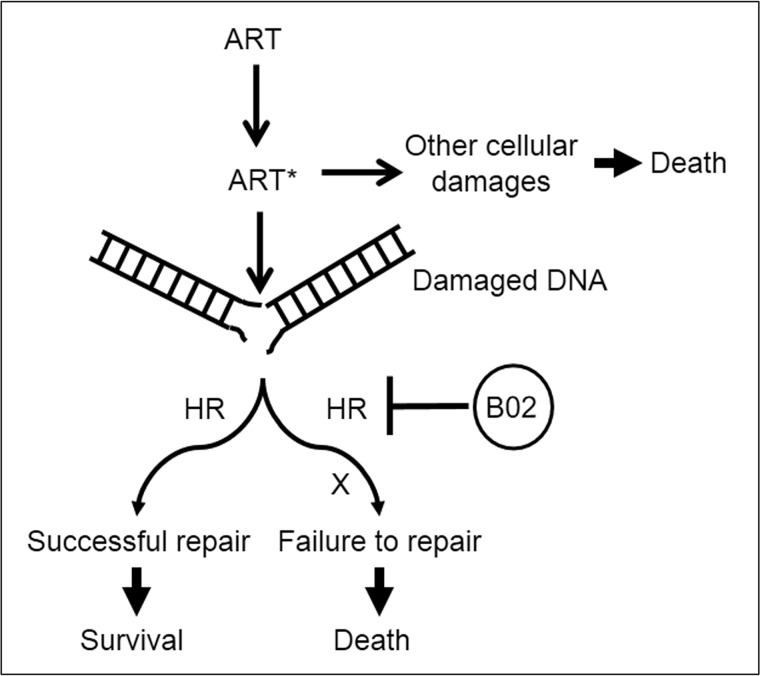
Malaria parasites repair DNA double-strand breaks (DSBs) primarily through homologous recombination (HR). Here, because the unrepaired DSBs lead to the death of the unicellular parasite Plasmodium falciparum, we investigated its recombinase, PfRad51, as a potential drug target. Undertaking an in silico screening approach, we identified a compound, B02, that docks to the predicted tertiary structure of PfRad51 with high affinity. B02 inhibited a drug-sensitive P. falciparum strain (3D7) and multidrug-resistant parasite (Dd2) in culture, with IC50 values of 8 and 3 μm, respectively. We found that B02 is more potent against these P. falciparum strains than against mammalian cell lines. Our findings also revealed that the antimalarial activity of B02 synergizes with those of two first-line malaria drugs, artemisinin (ART) and chloroquine (CQ), lowering the IC50 values of ART and CQ by 15- and 8-fold, respectively. Our results also provide mechanistic insights into the anti-parasitic activity of B02, indicating that it blocks the ATPase and strand-exchange activities of PfRad51 and abrogates the formation of PfRad51 foci on damaged DNA at chromosomal sites, probably by blocking homomeric interactions of PfRad51 proteins. The B02-mediated PfRad51 disruption led to the accumulation of unrepaired parasitic DNA and rendered parasites more sensitive to DNA-damaging agents, including ART. Our findings provide a rationale for targeting the Plasmodium DSB repair pathway in combination with ART. We propose that identification of a specific inhibitor of HR in Plasmodium may enable investigations of HR's role in Plasmodium biology, including generation of antigenic diversity.
Keywords: B02; DNA repair; PfRad51; artemisinin; comet assay; drug screening; homologous recombination; malaria; plasmodium; small-molecule inhibitor.
© 2019 Vydyam et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- World Health Organization. (2015) World Malaria Report 2015. World Health Organization, Geneva, Switzerland.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Research Materials
