

Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality
- PMID: 30936302
- PMCID: PMC6475421
- DOI: 10.1073/pnas.1818259116
Chemical synthesis rewriting of a bacterial genome to achieve design flexibility and biological functionality
Abstract
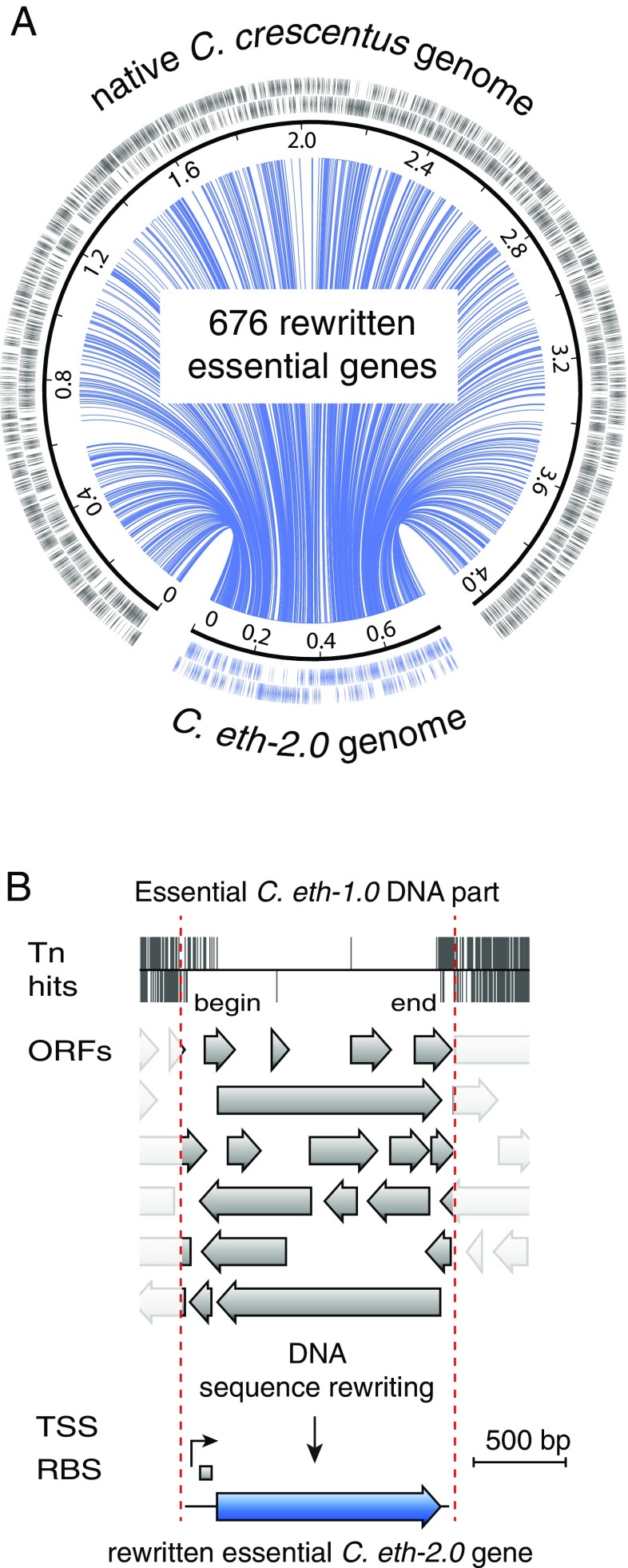
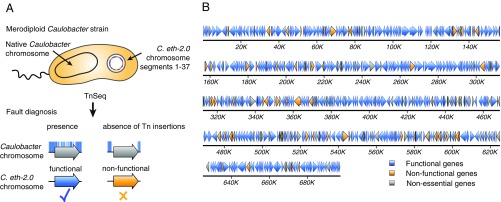
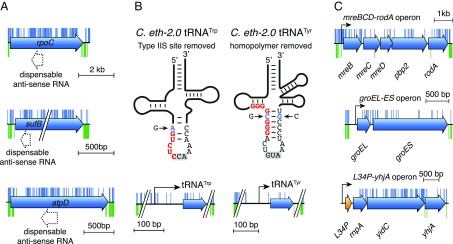
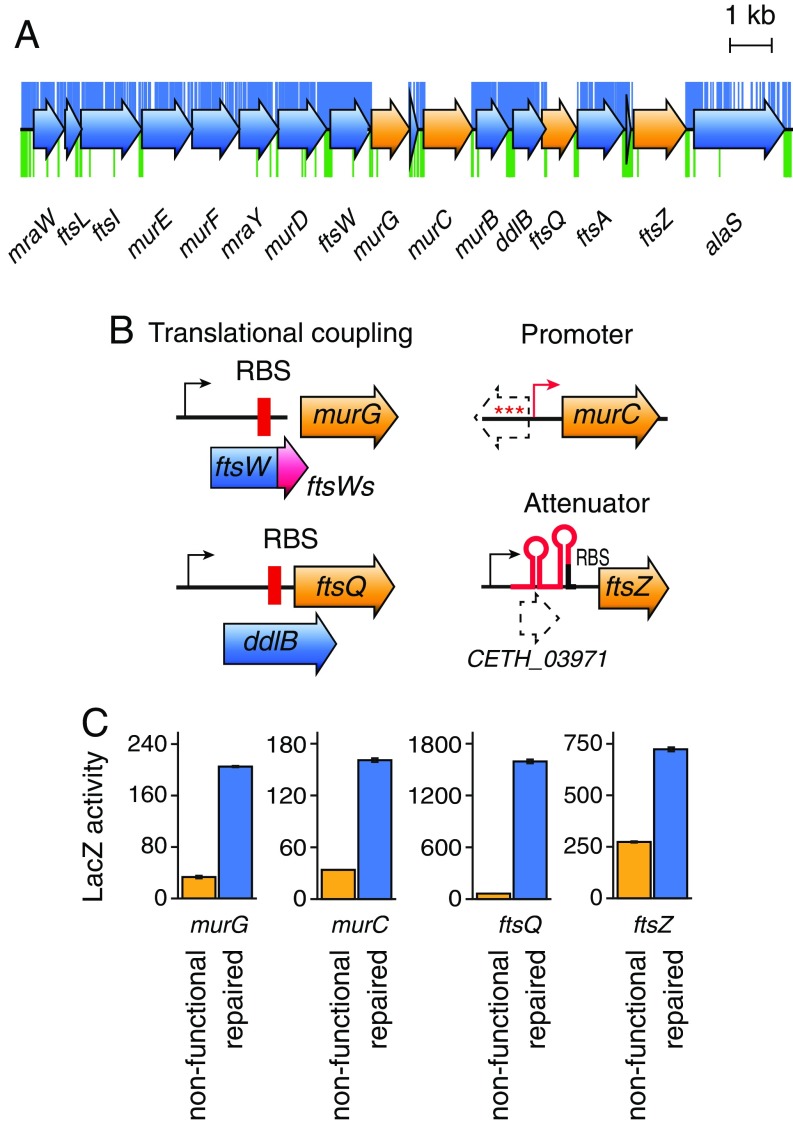
Understanding how to program biological functions into artificial DNA sequences remains a key challenge in synthetic genomics. Here, we report the chemical synthesis and testing of Caulobacter ethensis-2.0 (C. eth-2.0), a rewritten bacterial genome composed of the most fundamental functions of a bacterial cell. We rebuilt the essential genome of Caulobacter crescentus through the process of chemical synthesis rewriting and studied the genetic information content at the level of its essential genes. Within the 785,701-bp genome, we used sequence rewriting to reduce the number of encoded genetic features from 6,290 to 799. Overall, we introduced 133,313 base substitutions, resulting in the rewriting of 123,562 codons. We tested the biological functionality of the genome design in C. crescentus by transposon mutagenesis. Our analysis revealed that 432 essential genes of C. eth-2.0, corresponding to 81.5% of the design, are equal in functionality to natural genes. These findings suggest that neither changing mRNA structure nor changing the codon context have significant influence on biological functionality of synthetic genomes. Discovery of 98 genes that lost their function identified essential genes with incorrect annotation, including a limited set of 27 genes where we uncovered noncoding control features embedded within protein-coding sequences. In sum, our results highlight the promise of chemical synthesis rewriting to decode fundamental genome functions and its utility toward the design of improved organisms for industrial purposes and health benefits.
Keywords: Caulobacter crescentus; chemical genome synthesis; de novo DNA synthesis; genome rewriting; synonymous recoding.
Copyright © 2019 the Author(s). Published by PNAS.
Conflict of interest statement
Conflict of interest statement: Eidgenössische Technische Hochschule holds a patent application (WO2017085249A1) with M.C. and B.C. as inventors that covers functional testing of synthetic genomes. M.C. and B.C. hold shares from Gigabases Switzerland AG.
Figures

References
-
- Cello J, Paul AV, Wimmer E. Chemical synthesis of poliovirus cdna: Generation of infectious virus in the absence of natural template. Science. 2002;297:1016–1018. - PubMed
-
- Gibson DG, et al. Complete chemical synthesis, assembly, and cloning of a mycoplasma genitalium genome. Science. 2008;319:1215–1220. - PubMed
-
- Gibson DG, et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science. 2010;329:52–56. - PubMed
-
- Hutchison CA, et al. Design and synthesis of a minimal bacterial genome. Science. 2016;351:aad6253. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
