

Schilbach-Rott syndrome associated with 9q22.32q22.33 duplication, involving the PTCH1 gene
- PMID: 30936464
- PMCID: PMC6777447
- DOI: 10.1038/s41431-019-0385-6
Schilbach-Rott syndrome associated with 9q22.32q22.33 duplication, involving the PTCH1 gene
Abstract
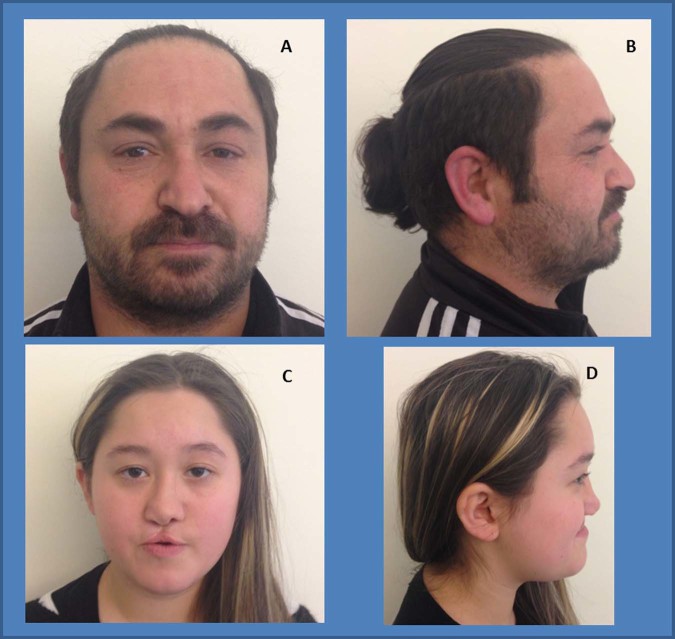
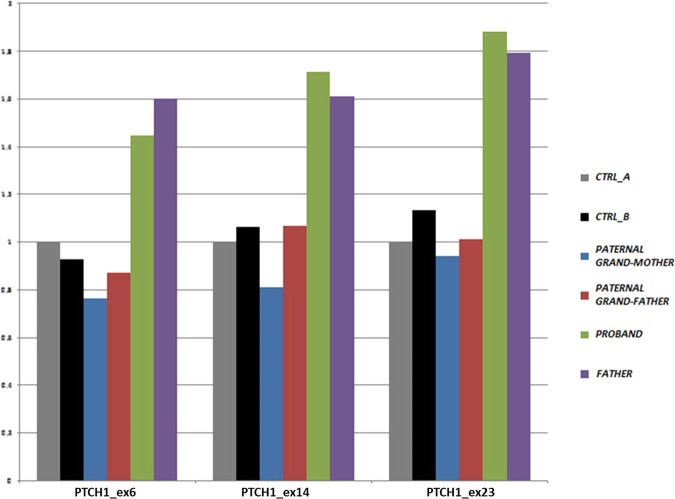
Schilbach-Rott syndrome (SRS, OMIM%164220) is a disorder of unknown aetiology that is characterised by hypotelorism, epichantal folds, cleft palate, dysmorphic face, hypospadia in males and mild mental retardation in some patients. To date, 5 families and 17 patients have exhibited this phenotype, and recurrence in two of these families suggests an autosomal dominant inheritance. SRS overlaps with a mild form of holoprosencephaly (HPE), but array-CGH analysis and sequencing of some HPE-related genes (SEPT9, SHH and TWIST) did not reveal any variants in at least one family. Herein, we investigated by array-CGH analysis a 11-year-old female patient and her father, both exhibiting the typical SRS phenotype, disclosing in the daughter-father couple the same microduplication of chromosome 9q22.32q22.33 [arr[hg19]9q22.32(98,049,611_98,049,636)x3,9q22.33 (99,301,483_99,301,508)x3], involving eight genes, including PTCH1. The duplication segregated with the disease, since it was not found in the healthy paternal grandparents of the proband. The gain-of-function variants of the PTCH1 gene are responsible for a mild form of HPE. This is the first genetic variant found in SRS. This finding reinforces the hypothesis that SRS belongs to the HPE clinical spectrum and suggests to perform array-CGH in patients with SRS phenotype and, if negative, to consider a potential benefit from sequencing of HPE-related genes.
Conflict of interest statement
Dr. Sallicandro's work has been funded by the “Baschirotto Institute of Rare Disease” (BIRD) Foundation. She has received compensation for carrying out a study on possible genetic causes of Schilbach–Rott syndrome. All other authors declare that they have no conflict of interest.
Figures


References
-
- Becerra-Solano LE, Casillas-Avila MP, Diaz-Rodriguez M, Nastasi-Catanese JA, Toscano-Flores JJ, Ramirez-Duenas ML. Schilbach-Rott syndrome in a third family: further delineation of an autosomal dominant trait. Genet Couns. 2007;18:317–23. - PubMed
-
- de Carvalho DR, Rossi NF, Schellini S, Moretti-Ferreira D, Richieri-Costa A. Schilbach-Rott/blepharofacio skeletal syndrome in a Brazilian patient. Am J MedGenet. 2008;146A:2134–7. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
