

Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation
- PMID: 30936548
- PMCID: PMC9533319
- DOI: 10.1038/s41591-019-0405-7
Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation
Erratum in
-
Author Correction: Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation.Nat Med. 2019 Dec;25(12):1948. doi: 10.1038/s41591-019-0663-4. Nat Med. 2019. PMID: 31664237
Abstract
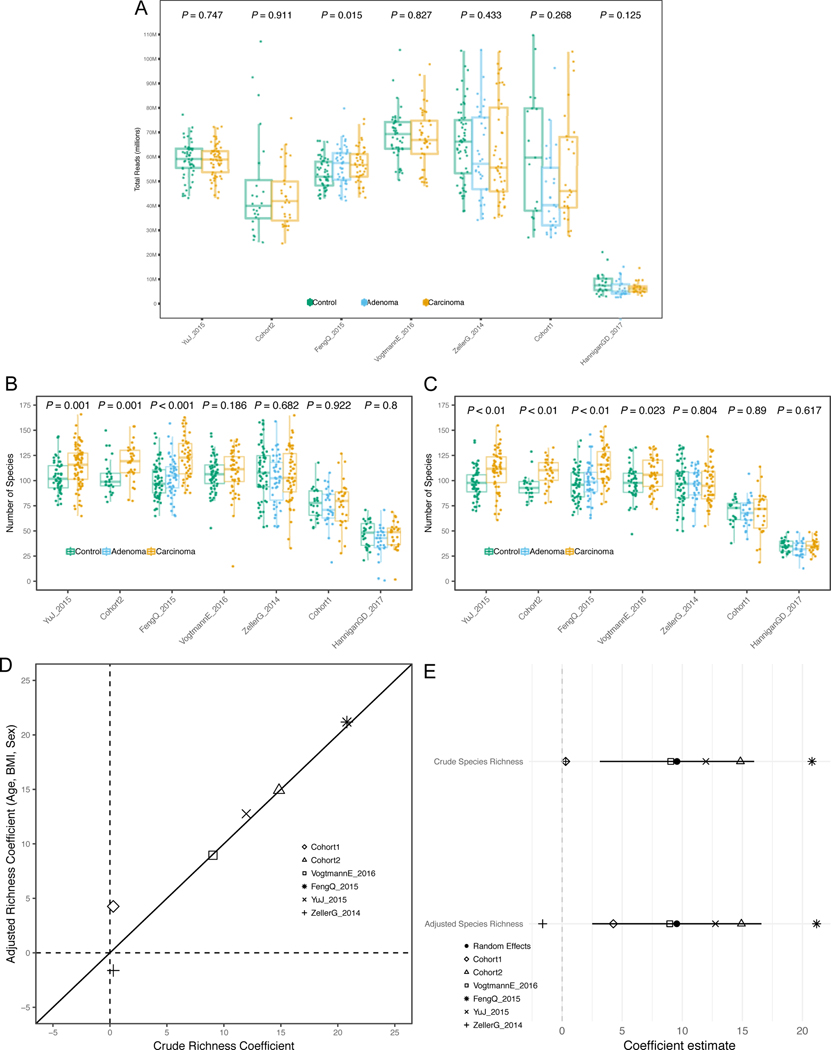
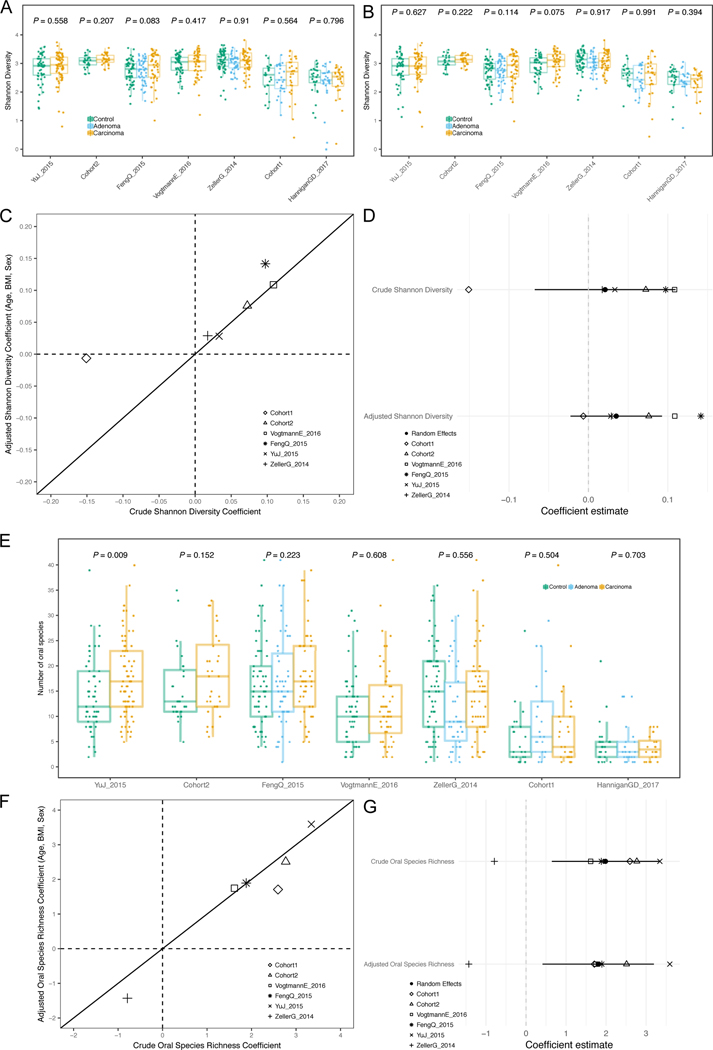
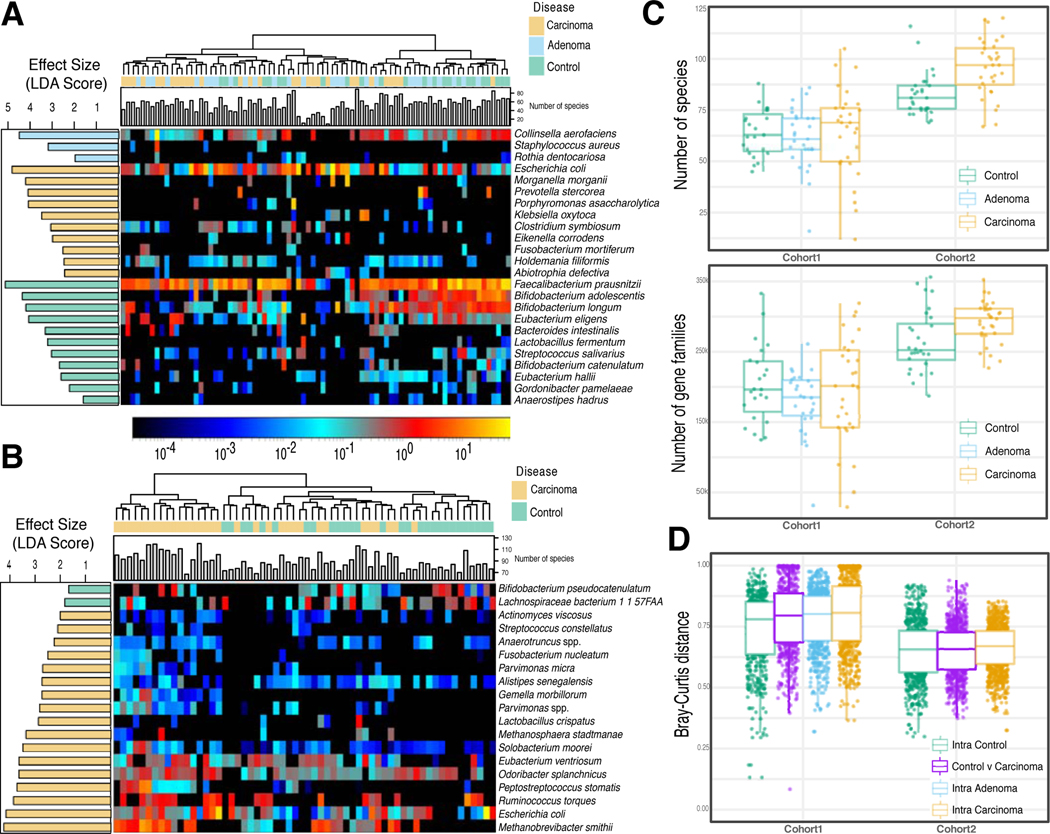
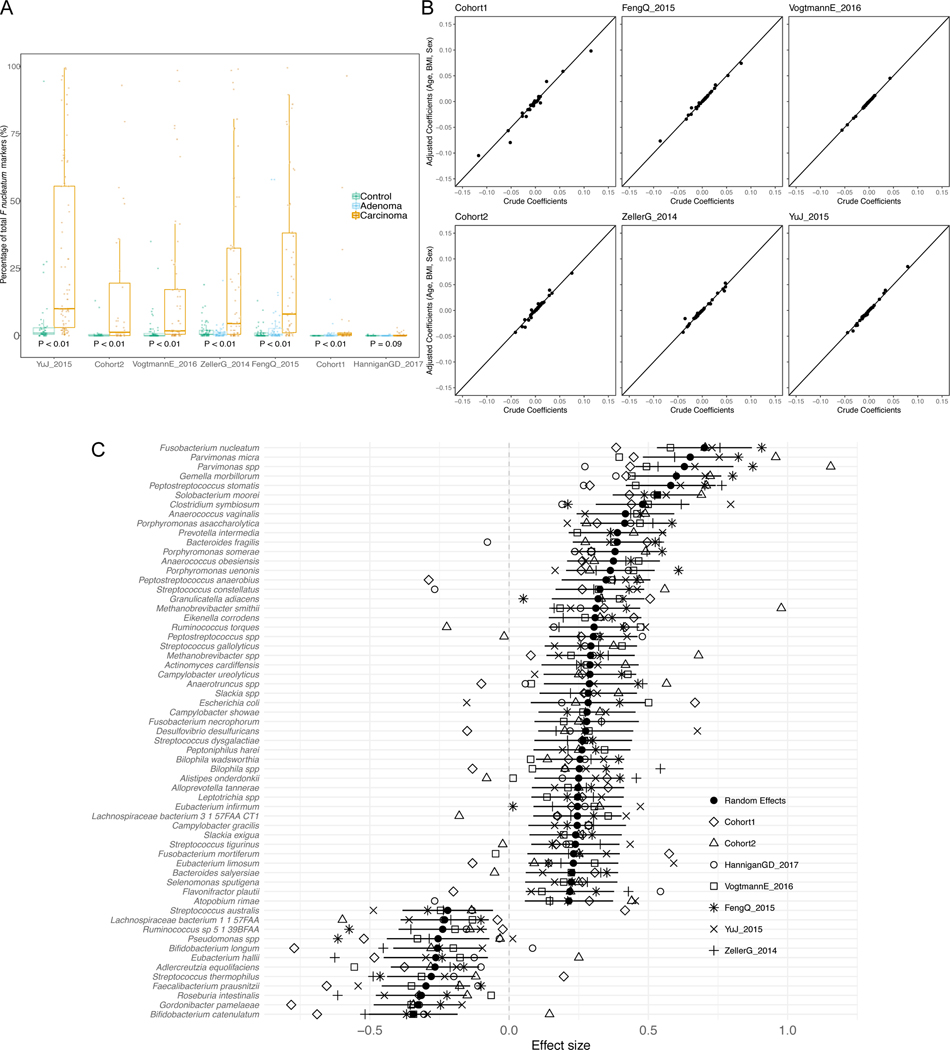
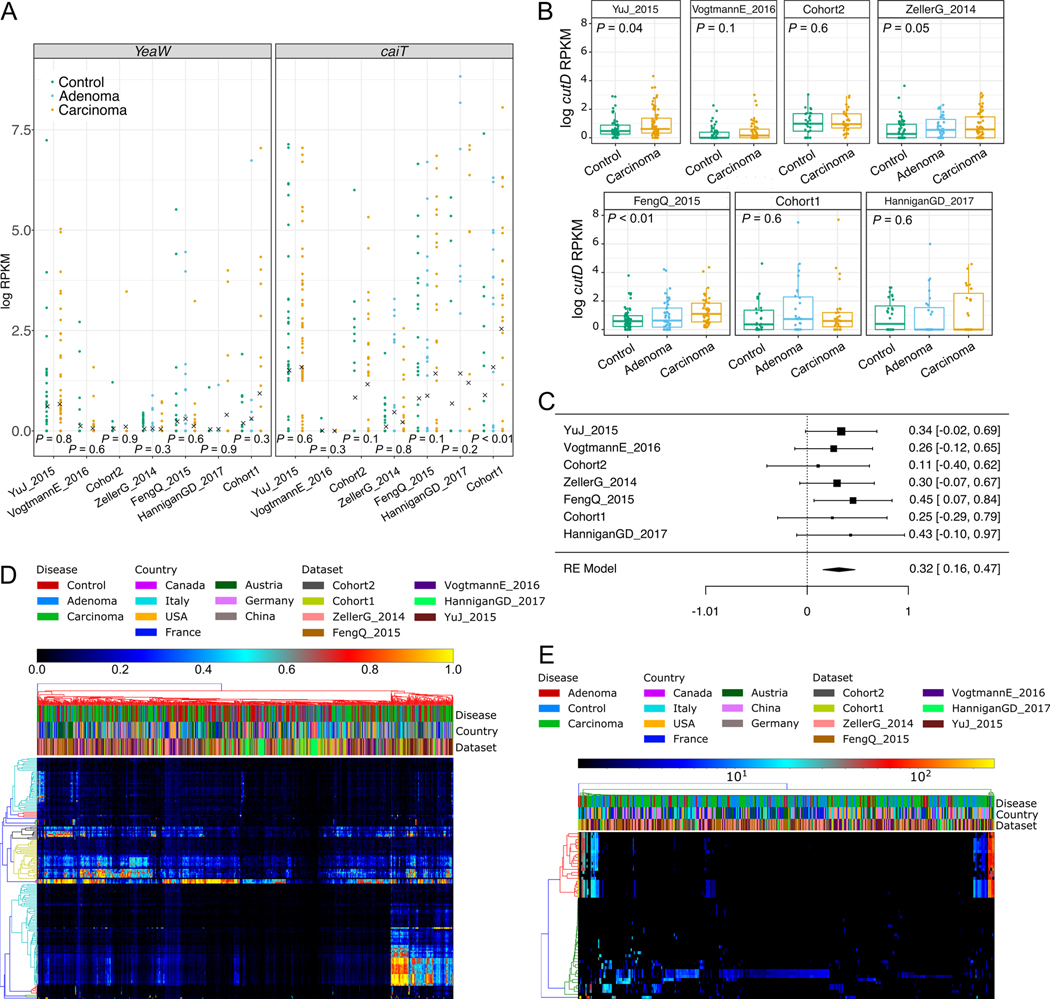
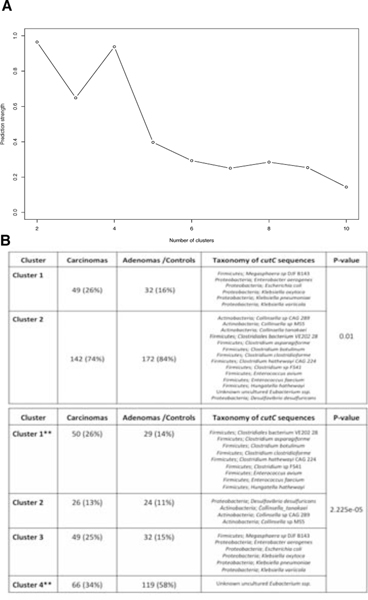
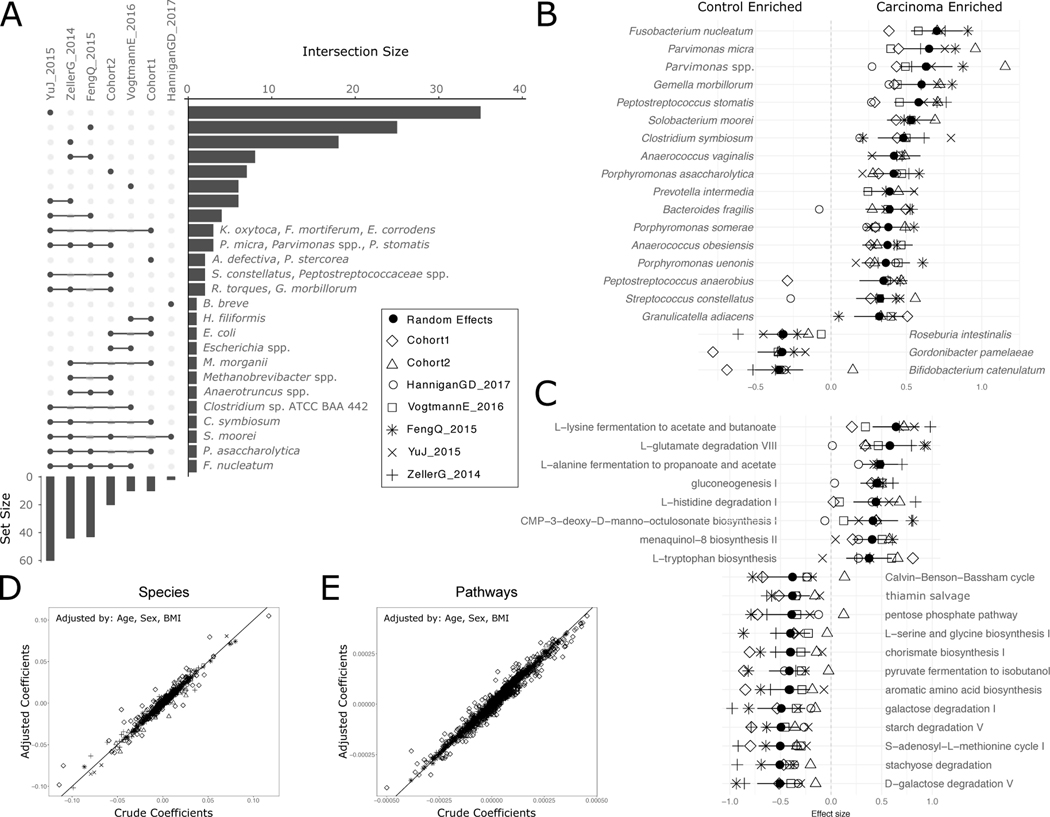
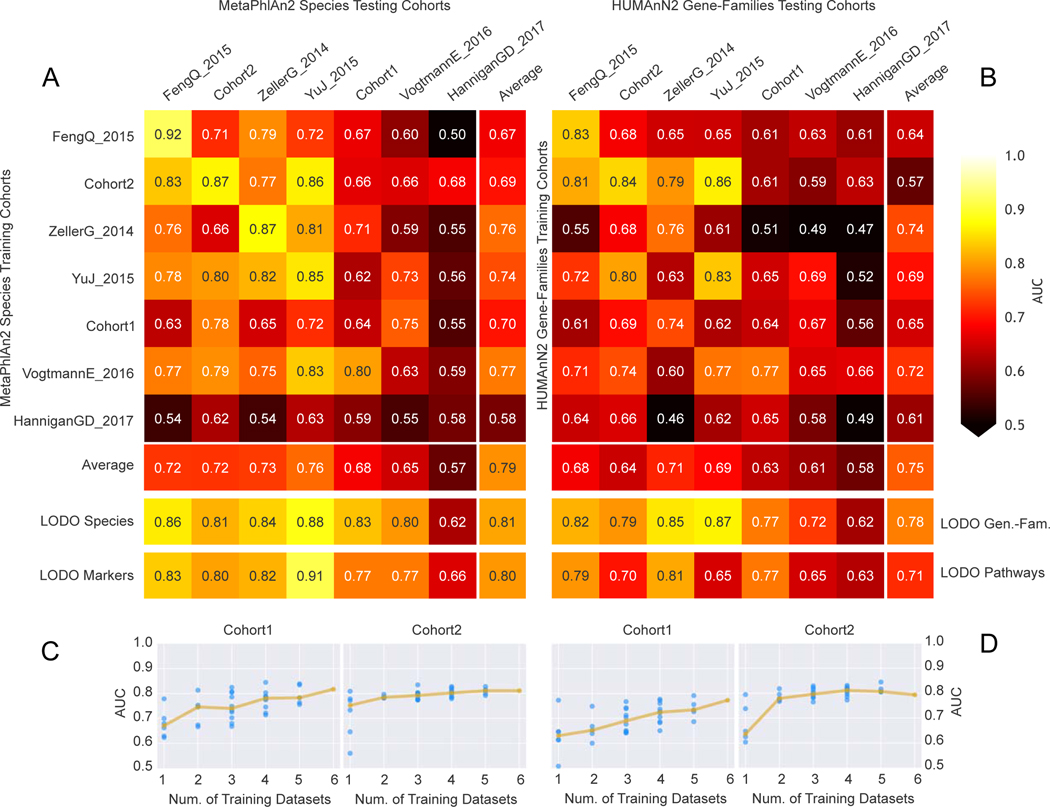
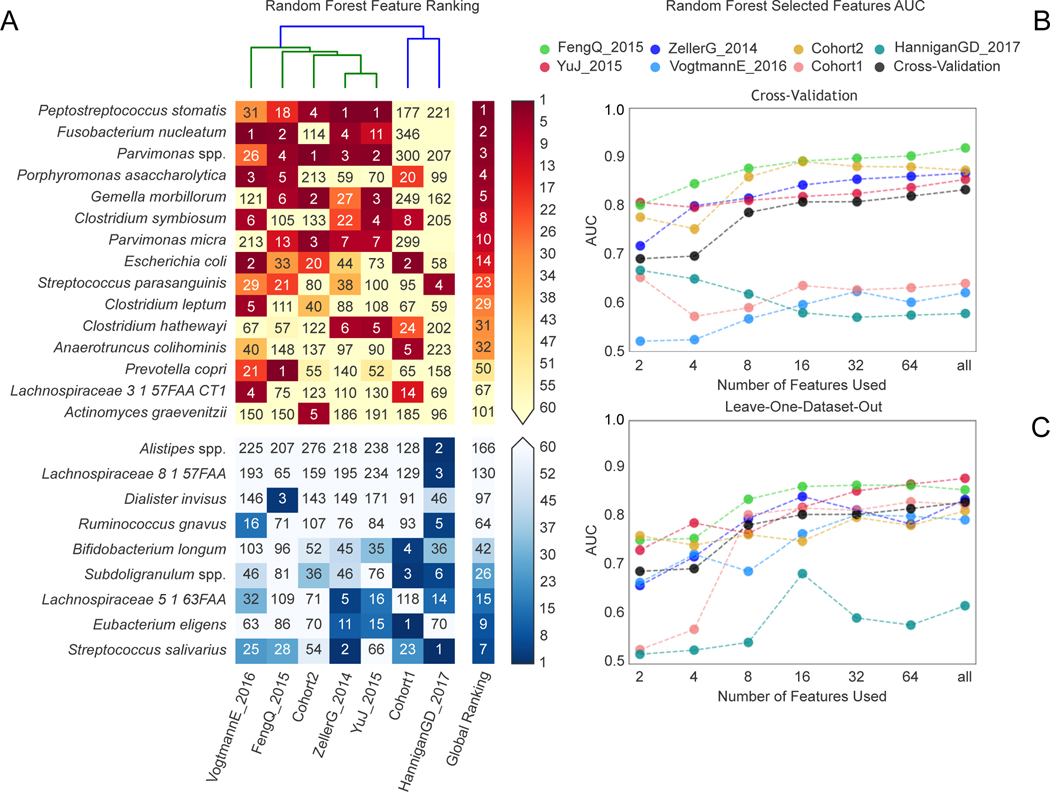
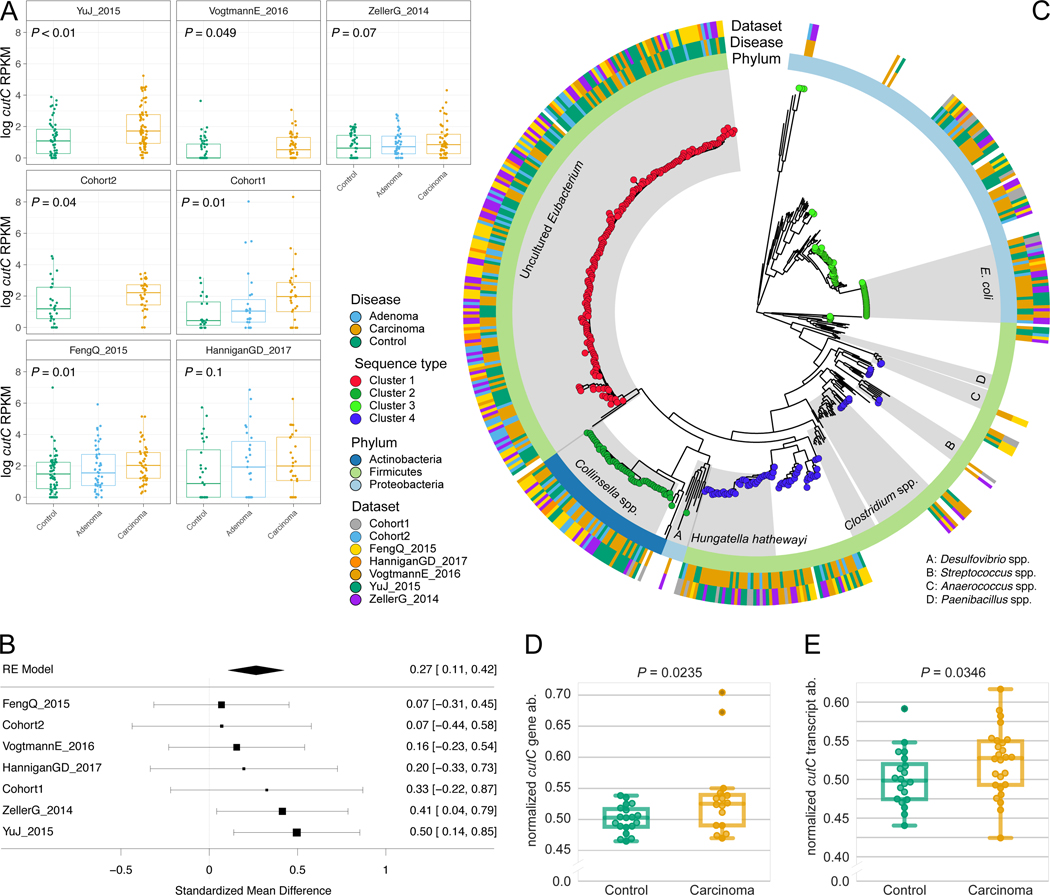
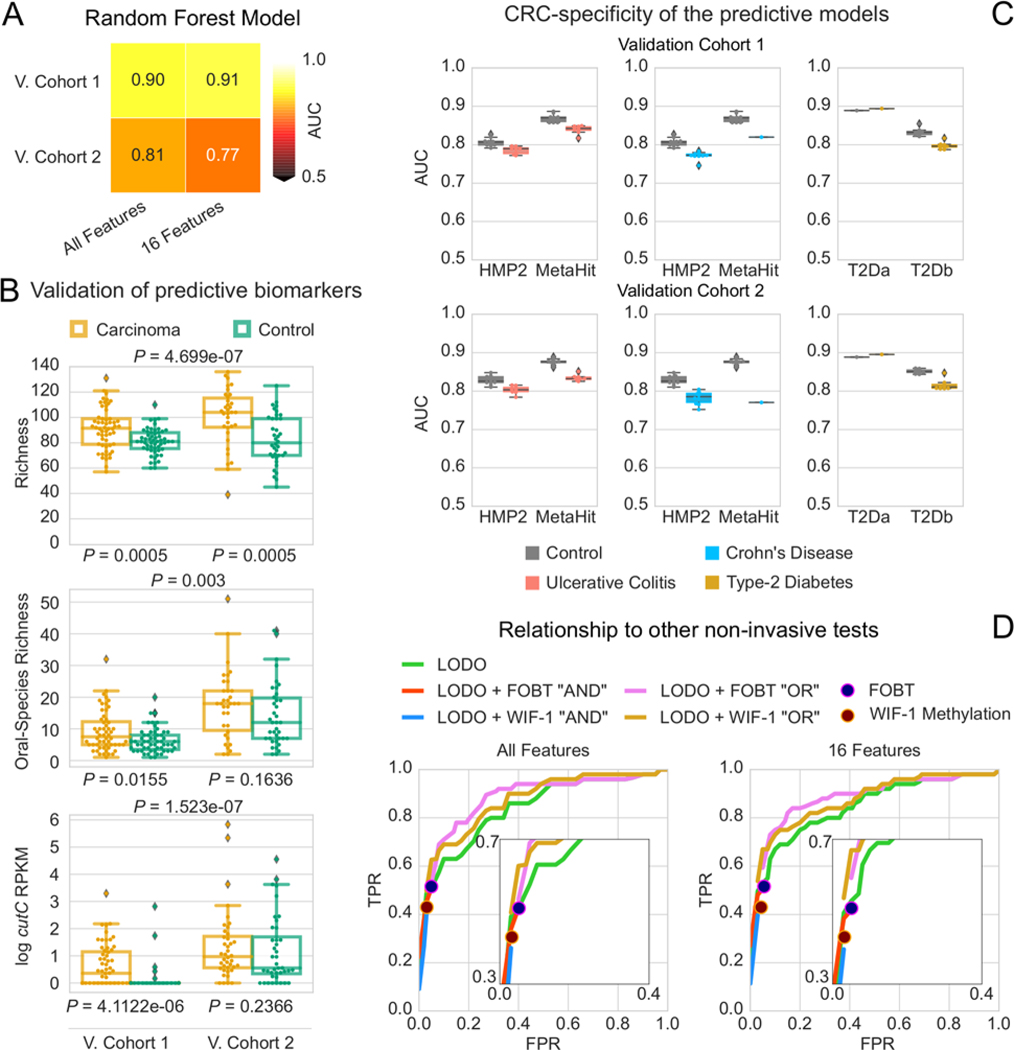
Several studies have investigated links between the gut microbiome and colorectal cancer (CRC), but questions remain about the replicability of biomarkers across cohorts and populations. We performed a meta-analysis of five publicly available datasets and two new cohorts and validated the findings on two additional cohorts, considering in total 969 fecal metagenomes. Unlike microbiome shifts associated with gastrointestinal syndromes, the gut microbiome in CRC showed reproducibly higher richness than controls (P < 0.01), partially due to expansions of species typically derived from the oral cavity. Meta-analysis of the microbiome functional potential identified gluconeogenesis and the putrefaction and fermentation pathways as being associated with CRC, whereas the stachyose and starch degradation pathways were associated with controls. Predictive microbiome signatures for CRC trained on multiple datasets showed consistently high accuracy in datasets not considered for model training and independent validation cohorts (average area under the curve, 0.84). Pooled analysis of raw metagenomes showed that the choline trimethylamine-lyase gene was overabundant in CRC (P = 0.001), identifying a relationship between microbiome choline metabolism and CRC. The combined analysis of heterogeneous CRC cohorts thus identified reproducible microbiome biomarkers and accurate disease-predictive models that can form the basis for clinical prognostic tests and hypothesis-driven mechanistic studies.
Figures
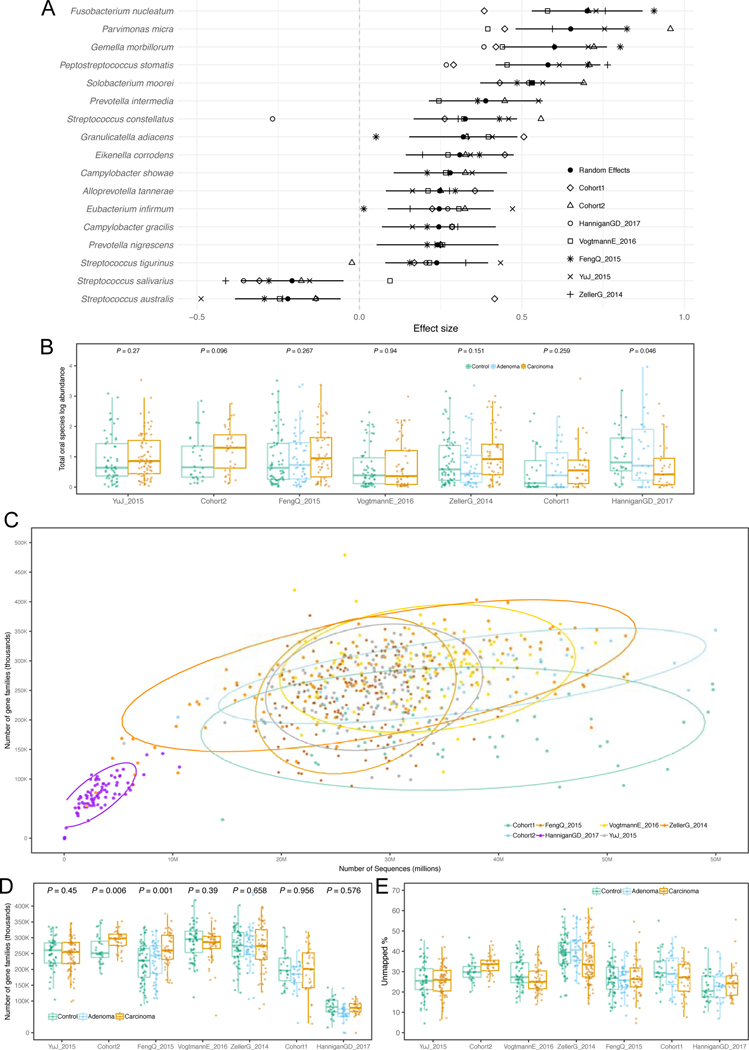
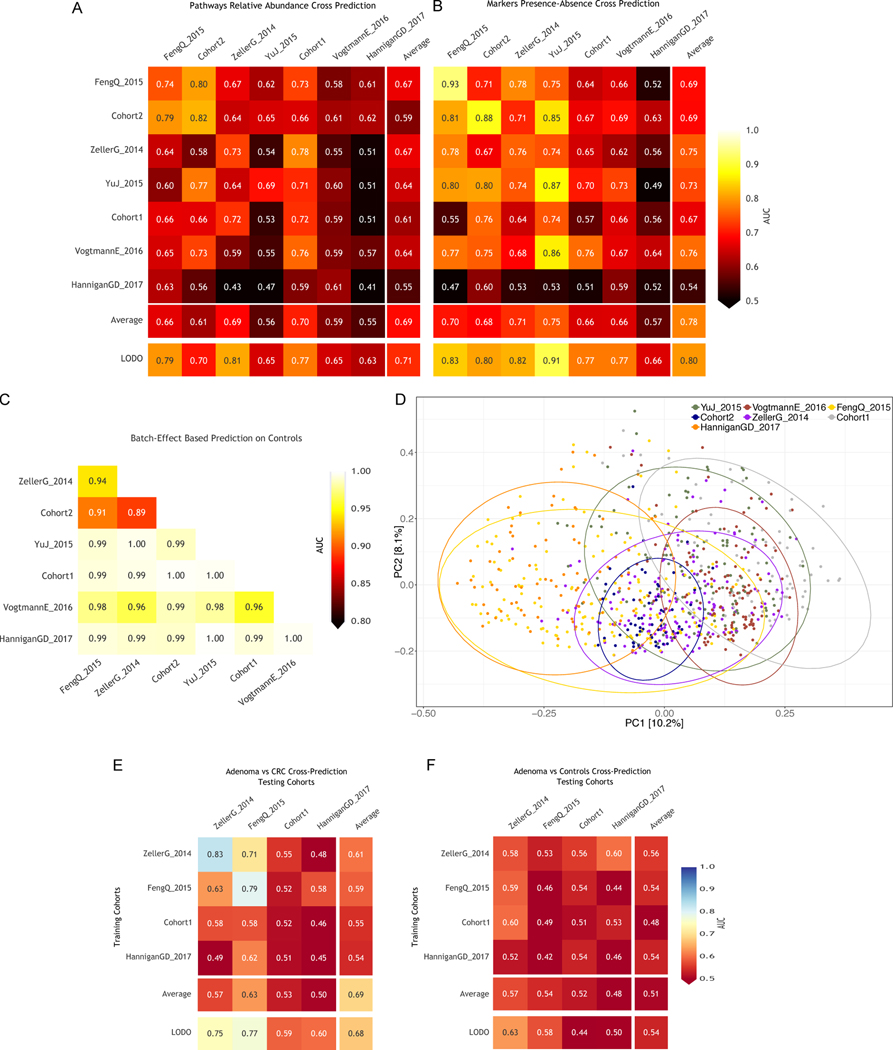
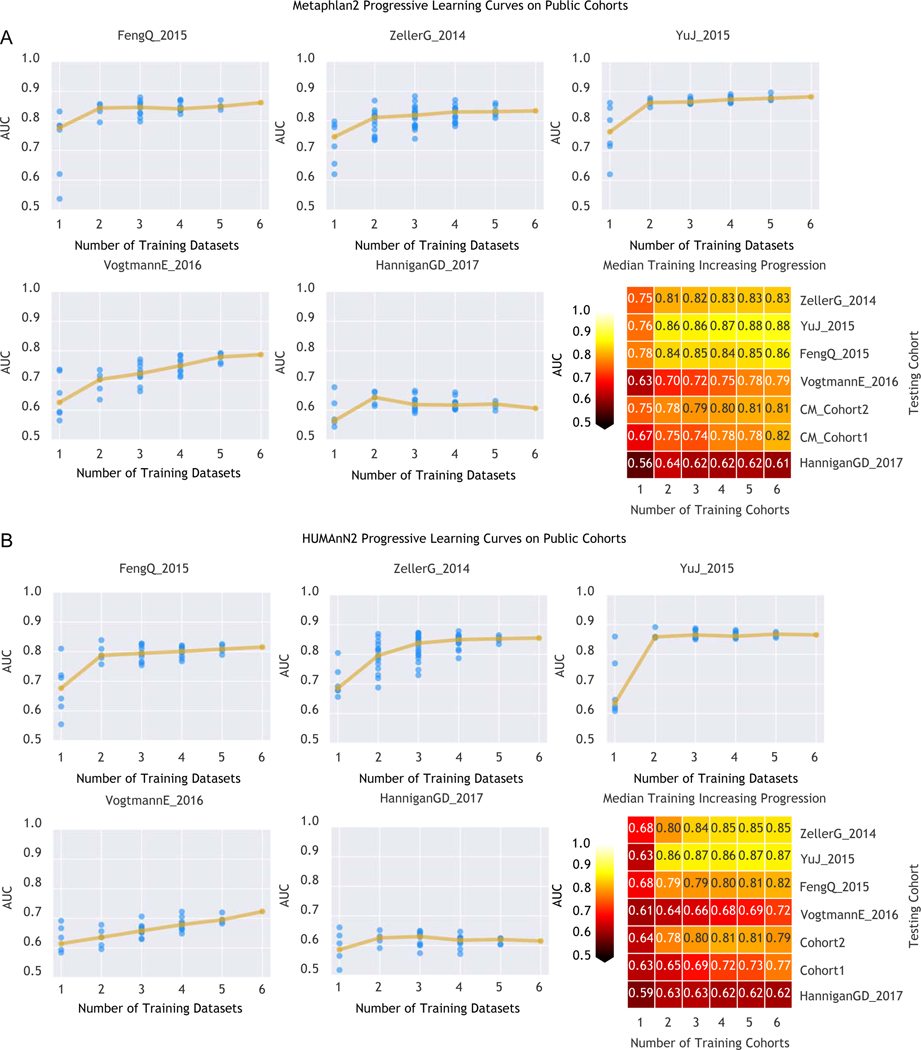
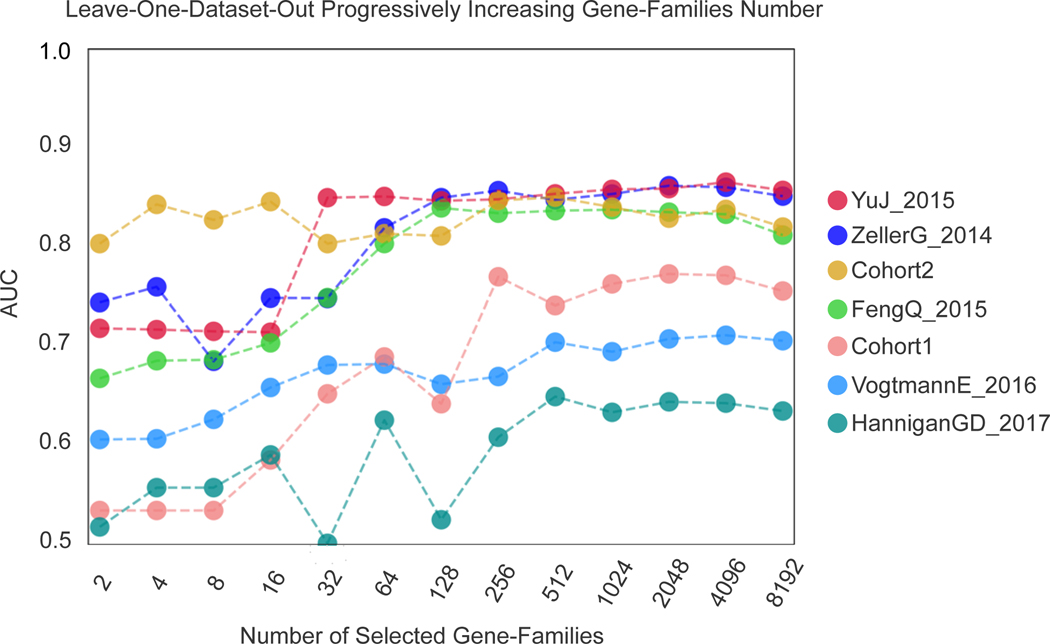
Comment in
-
Microbial signatures of colorectal cancer.Nat Rev Genet. 2019 Jun;20(6):318-319. doi: 10.1038/s41576-019-0126-2. Nat Rev Genet. 2019. PMID: 30971807 No abstract available.
References
-
- Ferlay J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–86 (2015). - PubMed
-
- Siegel R, Desantis C. & Jemal A. Colorectal cancer statistics, 2014. CA Cancer J. Clin 64, 104–117 (2014). - PubMed
-
- Frank C, Sundquist J, Yu H, Hemminki A. & Hemminki K. Concordant and discordant familial cancer: Familial risks, proportions and population impact. Int. J. Cancer 140, 1510–1516 (2017). - PubMed
-
- Foulkes WD Inherited susceptibility to common cancers. N. Engl. J. Med 359, 2143–2153 (2008). - PubMed
Methods-only References
-
- Truong DT et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903 (2015). - PubMed
-
- Breiman L. Random Forests. Mach. Learn 45, 5–32 (2001).
-
- Pedregosa F. et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830 (2011).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
