

An open resource for accurately benchmarking small variant and reference calls
- PMID: 30936564
- PMCID: PMC6500473
- DOI: 10.1038/s41587-019-0074-6
An open resource for accurately benchmarking small variant and reference calls
Abstract
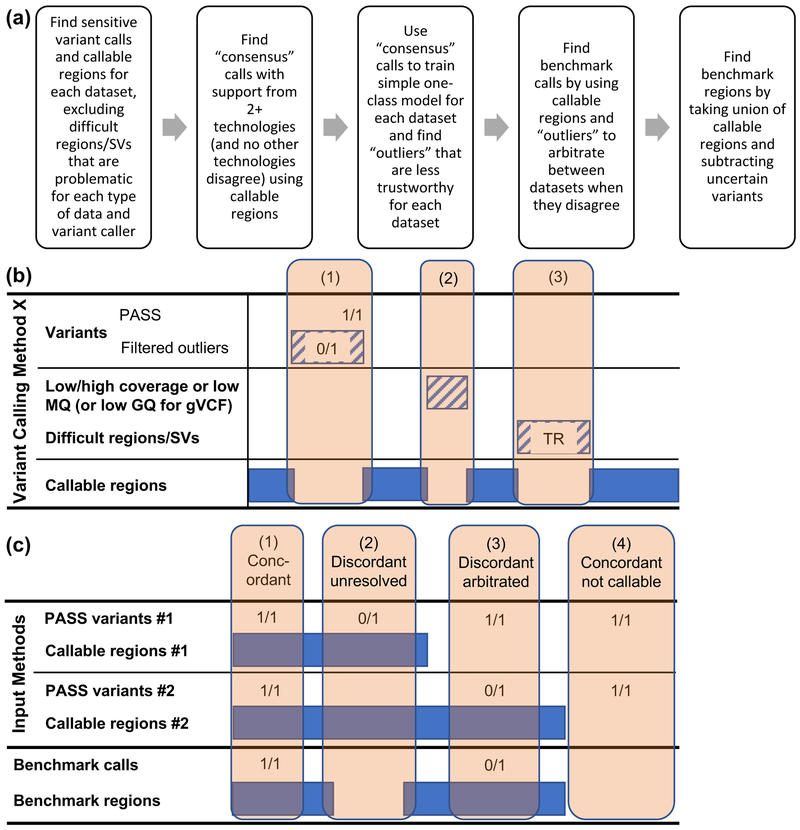
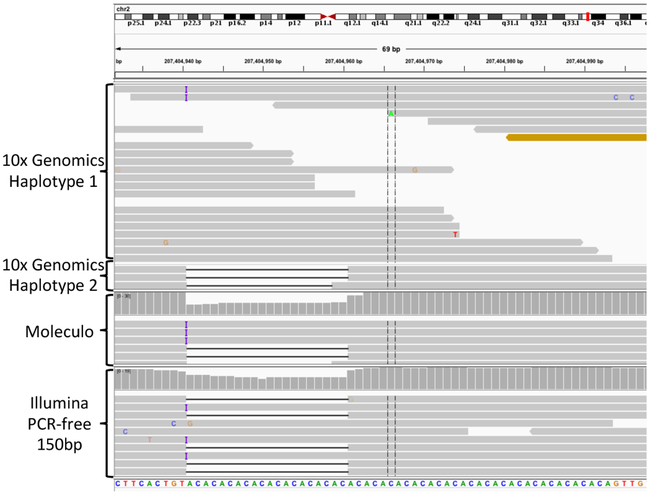
Benchmark small variant calls are required for developing, optimizing and assessing the performance of sequencing and bioinformatics methods. Here, as part of the Genome in a Bottle (GIAB) Consortium, we apply a reproducible, cloud-based pipeline to integrate multiple short- and linked-read sequencing datasets and provide benchmark calls for human genomes. We generate benchmark calls for one previously analyzed GIAB sample, as well as six genomes from the Personal Genome Project. These new genomes have broad, open consent, making this a 'first of its kind' resource that is available to the community for multiple downstream applications. We produce 17% more benchmark single nucleotide variations, 176% more indels and 12% larger benchmark regions than previously published GIAB benchmarks. We demonstrate that this benchmark reliably identifies errors in existing callsets and highlight challenges in interpreting performance metrics when using benchmarks that are not perfect or comprehensive. Finally, we identify strengths and weaknesses of callsets by stratifying performance according to variant type and genome context.
Figures
References
-
- Zook JM et al. Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–51 (2014). - PubMed
-
- Lincoln SE et al. A Systematic Comparison of Traditional and Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Genes in More Than 1000 Patients. J. Mol. Diagnostics 17, 533–544 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
