

Case Reports
doi: 10.1038/s41439-019-0046-x.
eCollection 2019.
A novel splicing mutation in SLC9A6 in a boy with Christianson syndrome
Affiliations
- PMID: 30937176
- PMCID: PMC6434044
- DOI: 10.1038/s41439-019-0046-x
Item in Clipboard
Case Reports
A novel splicing mutation in SLC9A6 in a boy with Christianson syndrome
Hum Genome Var.
.
Abstract
A loss of function mutation in SLC9A6 (Xq26.3) is responsible for Christianson syndrome in males. We identified a novel splicing mutation (NM_006359.2:c.1141-8C>A) of SLC9A6 in a seven-year-old boy with microcephaly, severe developmental delay, and intractable epilepsy. Functional analysis found multiple aberrant transcripts, none of which maintained the canonical open reading frame. Computer prediction tools, however, failed to detect all of the aberrant transcripts.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
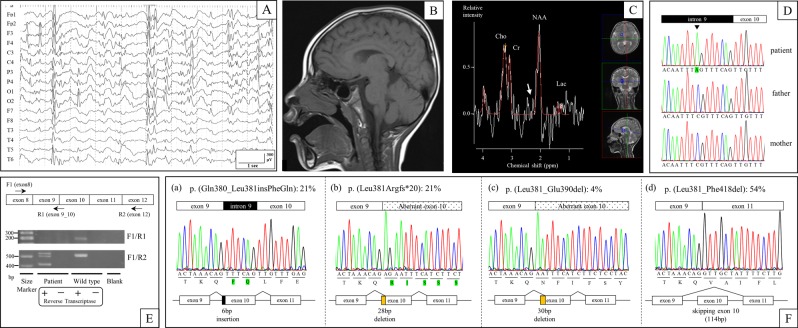
a EEG performed at 4 years showed focal epileptic discharge with generalization in multiple foci. b Brain MRI (T1 weighted sagittal) performed at 4 years did not show any abnormal findings. c Brain MRS in the basal ganglia performed at 4 years did not show any abnormal glutamate/glutamine peaks (white arrow). d The patient carried a de novo hemizygous SLC9A6 mutation (NM_006359.2:c.1141-8C>A) that was confirmed by Sanger sequencing. e RT-PCR analysis identified multiple aberrant transcripts but no canonical transcripts in the patient, while it identified only canonical transcripts in control DNA (wild type). f: Transcript variants in the patient. Twenty-one percent of transcripts included intronic 6-bp nucleotides (a), 21% excluded exonic 28-bp nucleotides (b), 4% excluded exonic 30-bp nucleotides (c), and 54% skipped exon 10 (d)
References
Publication types
LinkOut - more resources
Full Text Sources
