

Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants
- PMID: 30939815
- PMCID: PMC6520859
- DOI: 10.3390/v11040317
Development of a Versatile, Near Full Genome Amplification and Sequencing Approach for a Broad Variety of HIV-1 Group M Variants
Abstract
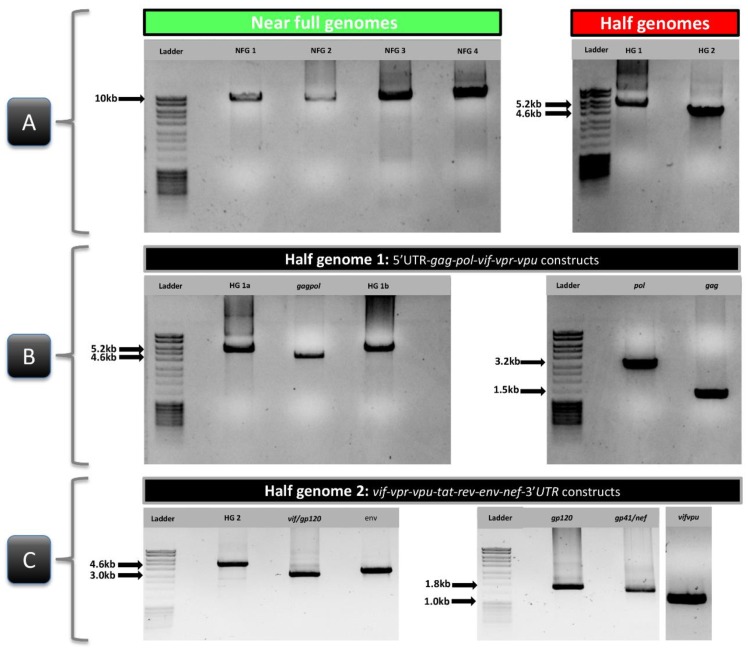
Near full genome sequencing (NFGS) of HIV-1 is required to assess the genetic composition of HIV-1 strains comprehensively. Population-wide, it enables a determination of the heterogeneity of HIV-1 and the emergence of novel/recombinant strains, while for each individual it constitutes a diagnostic instrument to assist targeted therapeutic measures against viral components. There is still a lack of robust and adaptable techniques for efficient NFGS from miscellaneous HIV-1 subtypes. Using rational primer design, a broad primer set was developed for the amplification and sequencing of diverse HIV-1 group M variants from plasma. Using pure subtypes as well as diverse, unique recombinant forms (URF), variable amplicon approaches were developed for NFGS comprising all functional genes. Twenty-three different genomes composed of subtypes A (A1), B, F (F2), G, CRF01_AE, CRF02_AG, and CRF22_01A1 were successfully determined. The NFGS approach was robust irrespective of viral loads (≥306 copies/mL) and amplification method. Third-generation sequencing (TGS), single genome amplification (SGA), cloning, and bulk sequencing yielded similar outcomes concerning subtype composition and recombinant breakpoint patterns. The introduction of a simple and versatile near full genome amplification, sequencing, and cloning method enables broad application in phylogenetic studies of diverse HIV-1 subtypes and can contribute to personalized HIV therapy and diagnosis.
Keywords: HIV-1 group M subtype-independent approach; Near full genome amplification and sequencing; bulk sequencing and cloning; rational primer design; single-genome amplification (SGA); third-generation sequencing (TGS).
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- HIV Circulating Recombinant Forms (CRFs) [(accessed on 27 January 2019)]; Available online: https://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
