

Discovering metabolic disease gene interactions by correlated effects on cellular morphology
- PMID: 30940487
- PMCID: PMC6531784
- DOI: 10.1016/j.molmet.2019.03.001
Discovering metabolic disease gene interactions by correlated effects on cellular morphology
Abstract
Objective: Impaired expansion of peripheral fat contributes to the pathogenesis of insulin resistance and Type 2 Diabetes (T2D). We aimed to identify novel disease-gene interactions during adipocyte differentiation.
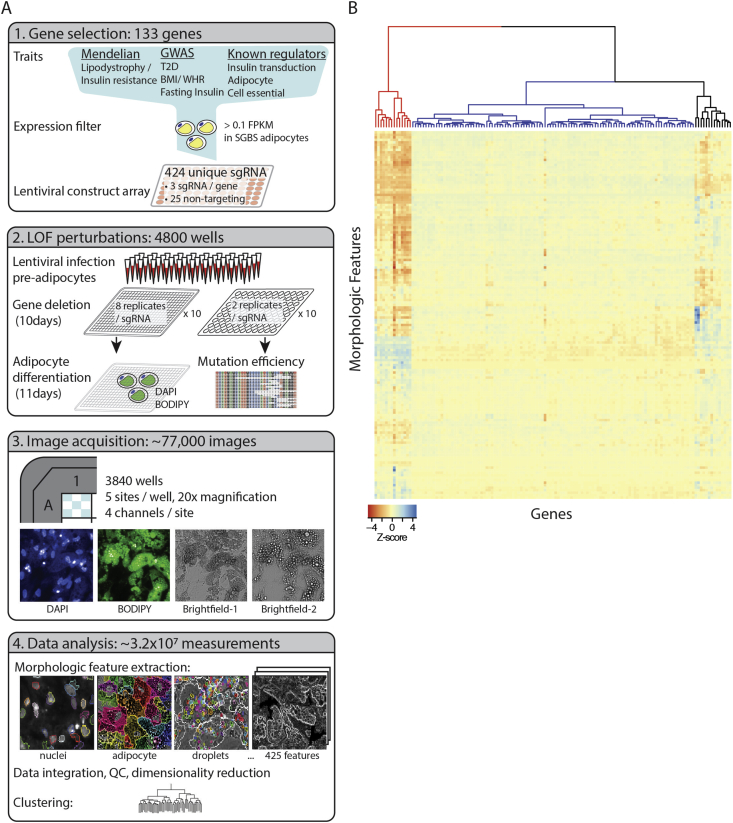
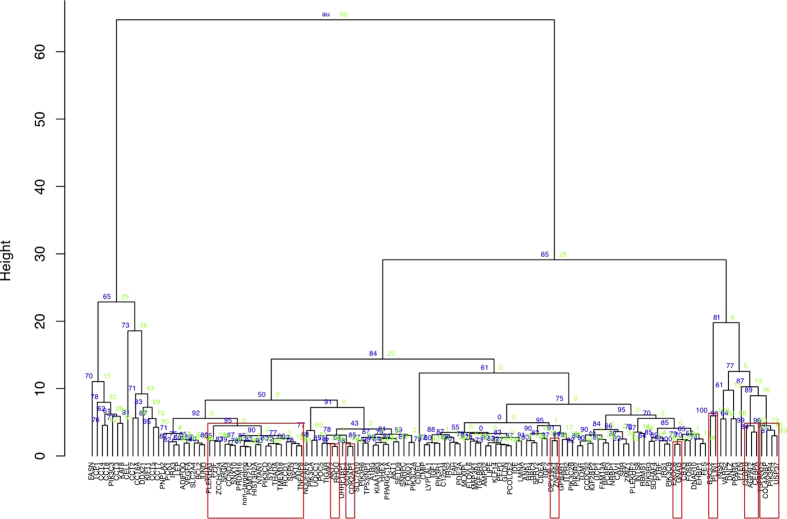
Methods: Genes in disease-associated loci for T2D, adiposity and insulin resistance were ranked according to expression in human adipocytes. The top 125 genes were ablated in human pre-adipocytes via CRISPR/CAS9 and the resulting cellular phenotypes quantified during adipocyte differentiation with high-content microscopy and automated image analysis. Morphometric measurements were extracted from all images and used to construct morphologic profiles for each gene.
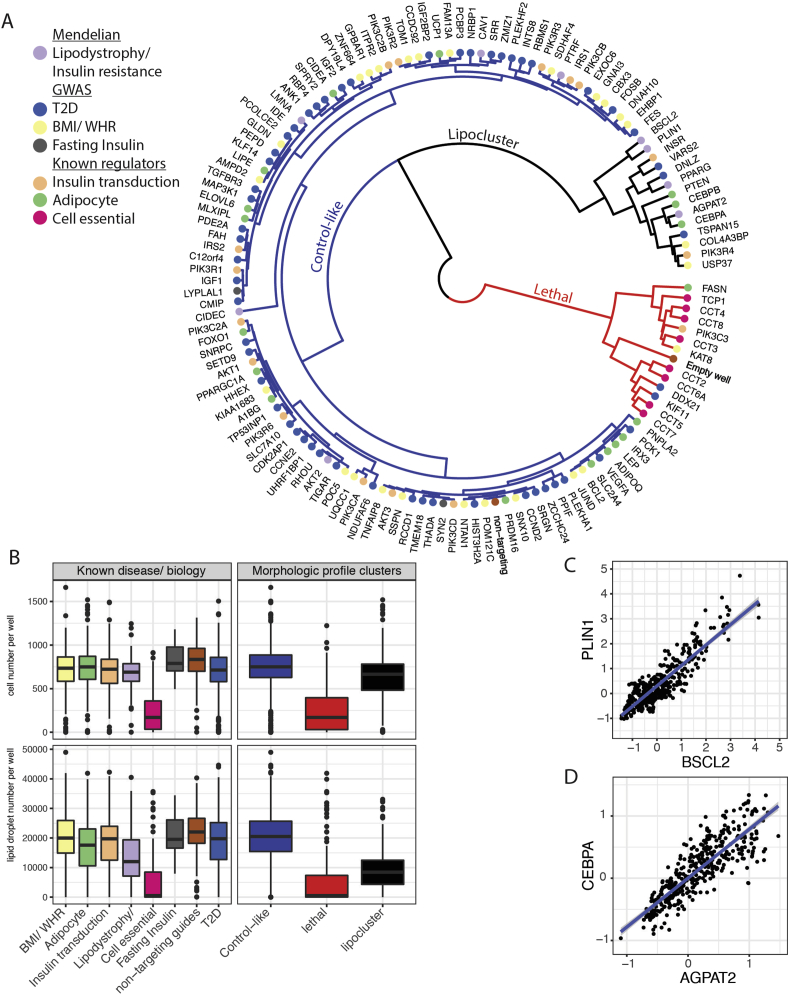
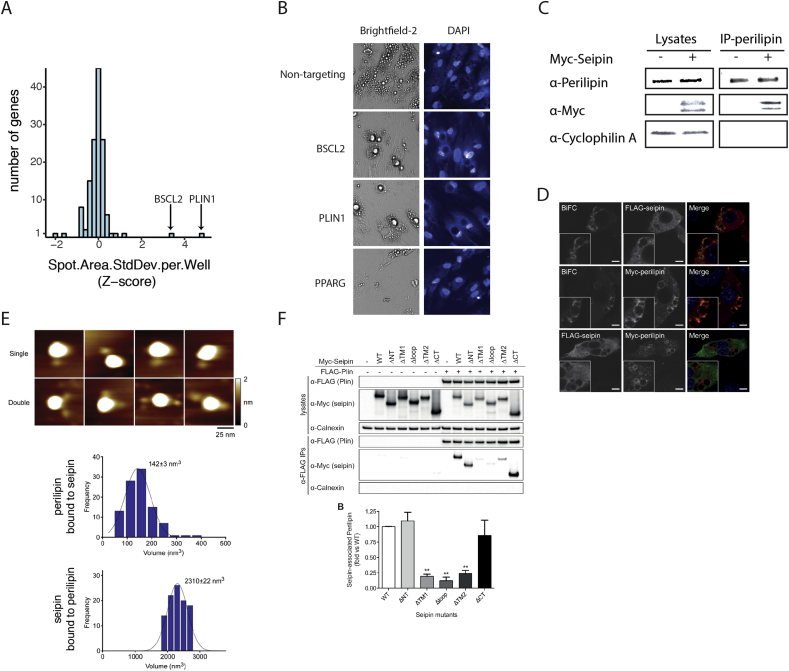
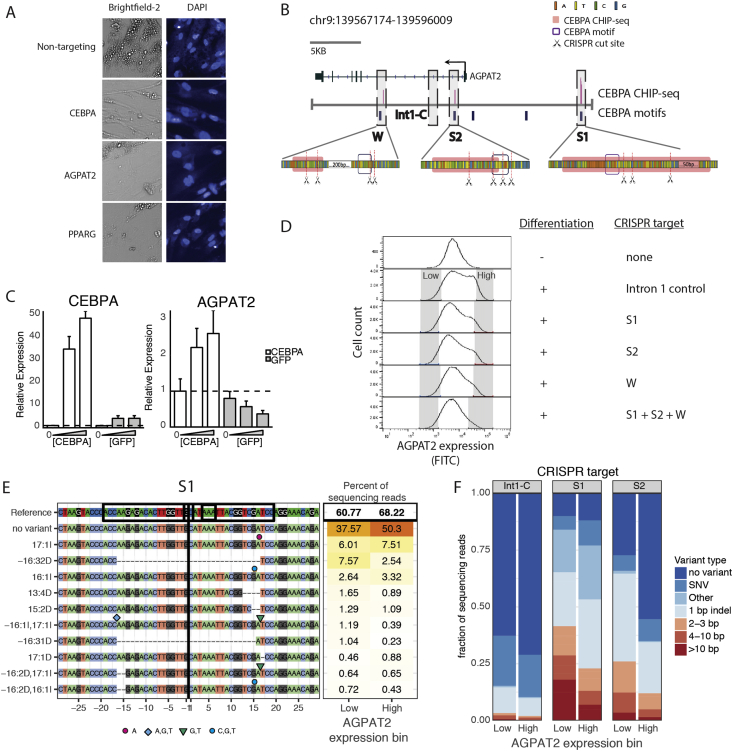
Results: Over 107 morphometric measurements were obtained. Clustering of the morphologic profiles accross all genes revealed a group of 14 genes characterized by decreased lipid accumulation, and enriched for known lipodystrophy genes. For two lipodystrophy genes, BSCL2 and AGPAT2, sub-clusters with PLIN1 and CEBPA identifed by morphological similarity were validated by independent experiments as novel protein-protein and gene regulatory interactions.
Conclusions: A morphometric approach in adipocytes can resolve multiple cellular mechanisms for metabolic disease loci; this approach enables mechanistic interrogation of the hundreds of metabolic disease loci whose function still remains unknown.
Keywords: Functional genomics; Gene discovery; Genetic screen; High content imaging; Insulin resistance; Lipodystrophy; Metabolic syndrome; Type 2 diabetes.
Copyright © 2019 The Authors. Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- Lamb J., Crawford E.D., Peck D., Modell J.W., Blat I.C., Wrobel M.J. The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. - PubMed
-
- Caicedo J.C., Singh S., Carpenter A.E. Applications in image-based profiling of perturbations. Current Opinion in Biotechnology. 2016;39:134–142. - PubMed
-
- Meijering E., Carpenter A.E., Peng H., Hamprecht F.A., Olivo-Marin J.-C. Imagining the future of bioimage analysis. Nature Biotechnology. 2016;34(12):1250–1255. - PubMed
