

Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology
- PMID: 30940804
- PMCID: PMC6445083
- DOI: 10.1038/s41467-019-09373-w
Whole genome sequencing of canids reveals genomic regions under selection and variants influencing morphology
Abstract
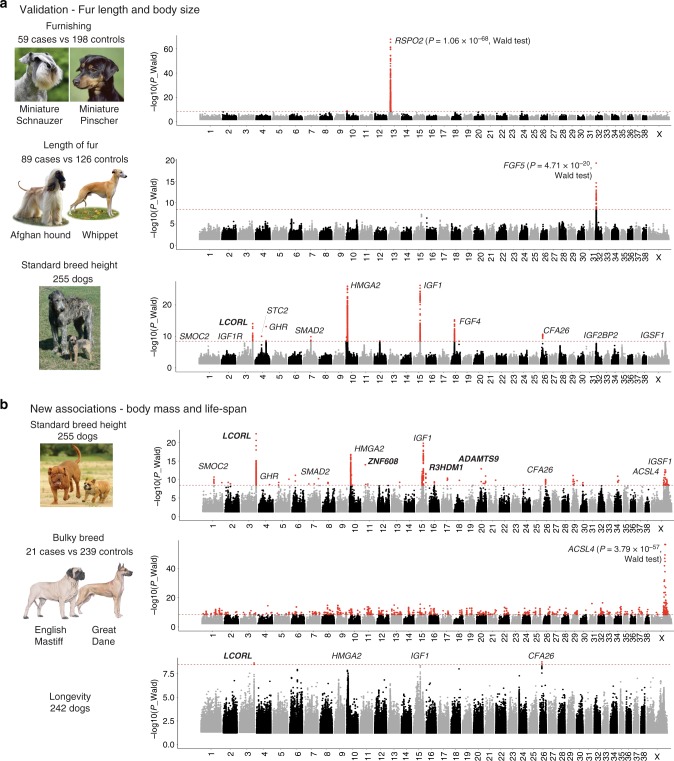
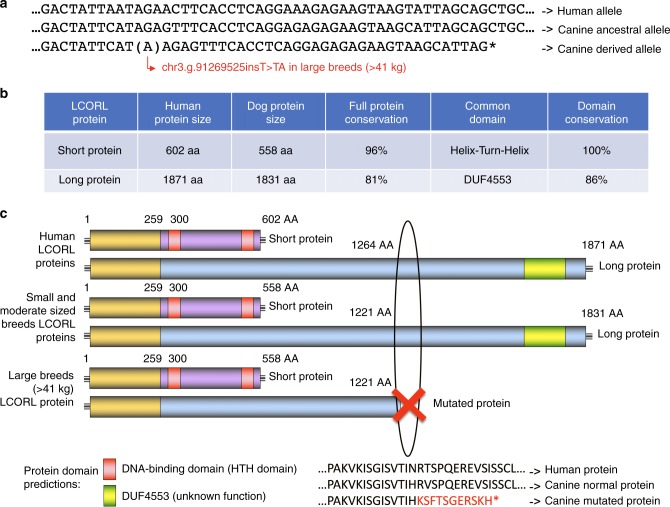
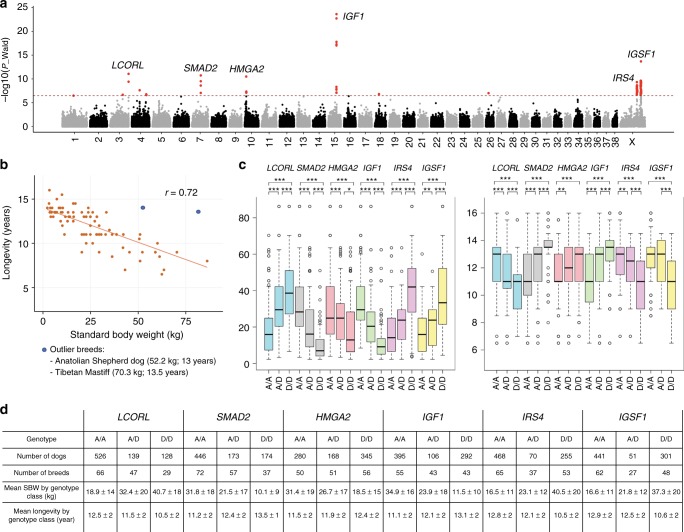
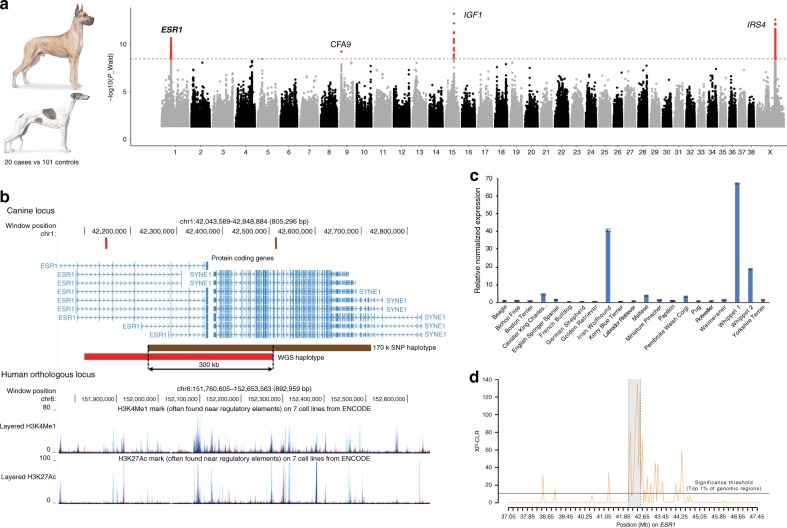
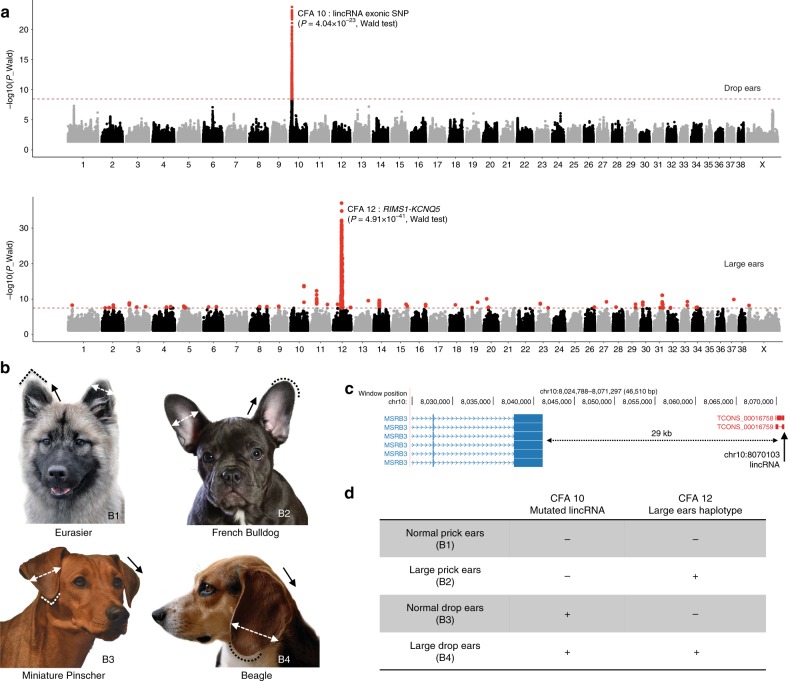
Domestic dog breeds are characterized by an unrivaled diversity of morphologic traits and breed-associated behaviors resulting from human selective pressures. To identify the genetic underpinnings of such traits, we analyze 722 canine whole genome sequences (WGS), documenting over 91 million single nucleotide and small indels, creating a large catalog of genomic variation for a companion animal species. We undertake both selective sweep analyses and genome wide association studies (GWAS) inclusive of over 144 modern breeds, 54 wild canids and a hundred village dogs. Our results identify variants of strong impact associated with 16 phenotypes, including body weight variation which, when combined with existing data, explain greater than 90% of body size variation in dogs. We thus demonstrate that GWAS and selection scans performed with WGS are powerful complementary methods for expanding the utility of companion animal systems for the study of mammalian growth and biology.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- American Kennel Club. The Complete Dog Book 20th edn (Ballantine Books, New York, NY, 2006).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
