

Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients
- PMID: 30941742
- PMCID: PMC6852597
- DOI: 10.1111/cge.13546
Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients
Abstract
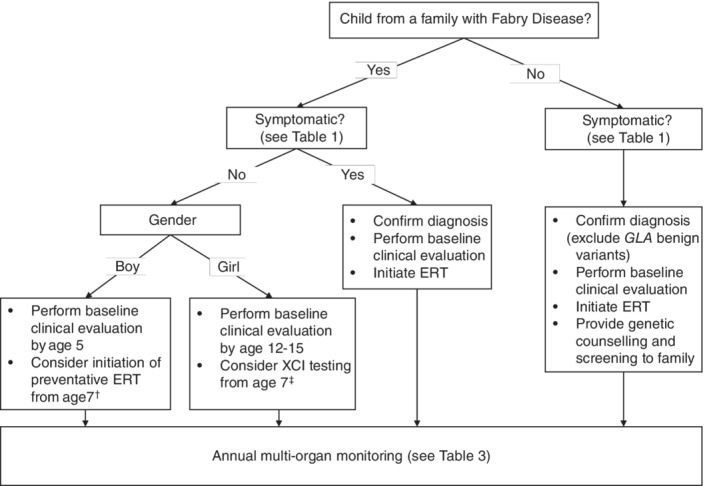
Fabry disease (FD), a rare X-linked disease, can be treated with bi-monthly infusion of enzyme replacement therapy (ERT) to replace deficient α-galactosidase A (AGAL-A). ERT reduces symptoms, improves quality of life (QoL), and improves clinical signs and biochemical markers. ERT initiation in childhood could slow or stop progressive organ damage. Preventative treatment of FD from childhood is thought to avoid organ damage in later life, prompting a French expert working group to collaborate and produce recommendations for treating and monitoring children with FD. Organ involvement should be assessed by age 5 for asymptomatic boys (age 12-15 for asymptomatic girls), and immediately for children diagnosed via symptoms. The renal, cardiac, nervous and gastrointestinal systems should be assessed, as well as bone, skin, eyes, hearing, and QoL. The plasma biomarker globotriaosylsphingosine is also useful. ERT should be considered for symptomatic boys and girls with neuropathic pain, pathological albuminuria (≥3 mg/mmol creatinine), severe GI involvement and abdominal pain or cardiac involvement. ERT should be considered for asymptomatic boys from the age of 7. Organ involvement should be treated as needed. Early diagnosis and management of FD represents a promising strategy to reduce organ damage, morbidity and premature mortality in adulthood.
Keywords: Fabry disease; children; diagnosis; enzyme replacement therapy; management; paediatric; treatment.
© 2019 The Authors. Clinical Genetics published by John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
D.P.G. is a consultant for Amicus, Sanofi‐Genzyme and Shire. He has received speaker's honoraria from Amicus, Sanofi‐Genzyme and Shire. A.F. has received speaker's and consultation honoraria from Amicus Therapeutics, Biomarin and Sanofi‐Genzyme. S.D. has received speaker's honoraria from Sanofi‐Genzyme, Shire and Amicus. M.T. received travel grants and speaker's honoraria from Sanofi‐Genzyme. P.P. received consultant honoraria from Sanofi‐Genzyme. M.F. received travel grants from Genzyme, Shire and Alexion. S.R. reports no conflict of interest. G.D. has received travel grants and inscription fees from Sanofi‐Genzyme and Alexion Pharma France. D.L. is a consultant for Sanofi‐Genzyme and has received travel grants and speaker's honoraria from Amicus, Biomarin, Shire and Sanofi‐Genzyme.
Figures
References
-
- Brady RO, Gal AE, Bradley RM, Martensson E, Warshaw AL, Laster L. Enzymatic defect in Fabry's disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163‐1167. - PubMed
-
- Ries M, Ramaswami U, Parini R, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162(11):767‐772. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
