

Characteristics of Pompe disease in China: a report from the Pompe registry
- PMID: 30943998
- PMCID: PMC6448270
- DOI: 10.1186/s13023-019-1054-0
Characteristics of Pompe disease in China: a report from the Pompe registry
Abstract
Background: Pompe disease is a rare, progressive, autosomal recessive lysosomal storage disorder caused by mutations in the acid α-glucosidase gene. This is the first report of Chinese patients from the global Pompe Registry. Chinese patients enrolled in the Registry ( ClinicalTrials.gov , NCT00231400) between Jan 2013 and 2 Sep 2016 with late onset Pompe disease (LOPD; presentation after 12 months of age or presentation at ≤12 months without cardiomyopathy) were included. Data analyses were descriptive.
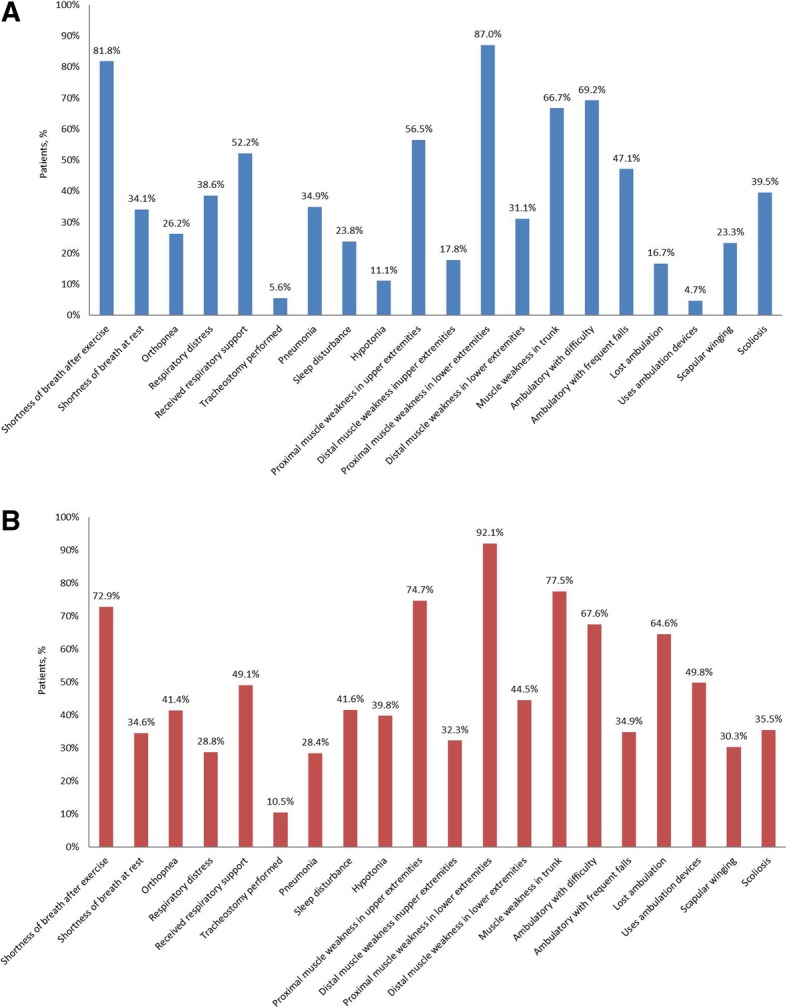
Results: Of the 59 Chinese patients included, 86.4% had never received enzyme replacement therapy (ERT). The age at symptom onset and diagnosis was 14.9 (12.35) and 22.1 (10.08) years, which is younger than previous reports of LOPD patients from the rest of the world (28.4 [18.86] and 34.9 [20.03], respectively). The most common diagnosis methods were enzyme assay (79.7%) and/or DNA analysis (61.0%). Of the 36 patients diagnosed using DNA analysis, 31 had standardized variant data and among these patients the most common mutations were c.2238G > C (n = 18, 58.1%) and c.2662G > T (n = 5, 16.1%). Chinese LOPD patients appeared to have worse lung function versus patients from the rest of the world, indicated by lower forced vital capacity (37.2 [14.00]% vs. 63.5 [26.71]%) and maximal expiratory and inspiratory pressure (27.9 [13.54] vs. 51.0 [38.66] cm H2O, and 29.4 [12.04] vs. 70.5 [52.78] cm H2O).
Conclusions: Compared with patients from the rest of the world, Chinese patients with LOPD appeared to have younger age at symptom onset and diagnosis, lower lung function, and the majority had not received ERT. The most common mutations were c.2238G > C and c.2662G > T.
Keywords: China; Late onset Pompe disease; Pompe disease; Pompe registry.
Conflict of interest statement
Ethics approval and consent to participate
Approval from a local institutional review board/ethics committee was required for inclusion in the Registry. Patient participation is voluntary, and written informed consent is obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors and Sanofi Genzyme declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. The metabolic and molecular bases of inherited disease. 2001;3:3389–3420.
-
- Ausems MG, Verbiest J, Hermans MP, Kroos MA, Beemer FA, Wokke JH, Sandkuijl LA, Reuser AJ, van der Ploeg AT. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999;7:713–716. doi: 10.1038/sj.ejhg.5200367. - DOI - PubMed
-
- Martiniuk F, Chen A, Mack A, Arvanitopoulos E, Chen Y, Rom WN, Codd WJ, Hanna B, Alcabes P, Raben N, Plotz P. Carrier frequency for glycogen storage disease type II in New York and estimates of affected individuals born with the disease. Am J Med Genet. 1998;79:69–72. doi: 10.1002/(SICI)1096-8628(19980827)79:1<69::AID-AJMG16>3.0.CO;2-K. - DOI - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
