

Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models
- PMID: 30944222
- PMCID: PMC6475399
- DOI: 10.1073/pnas.1818281116
Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models
Abstract
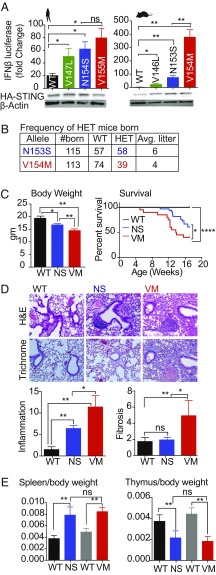
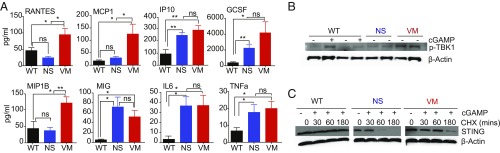
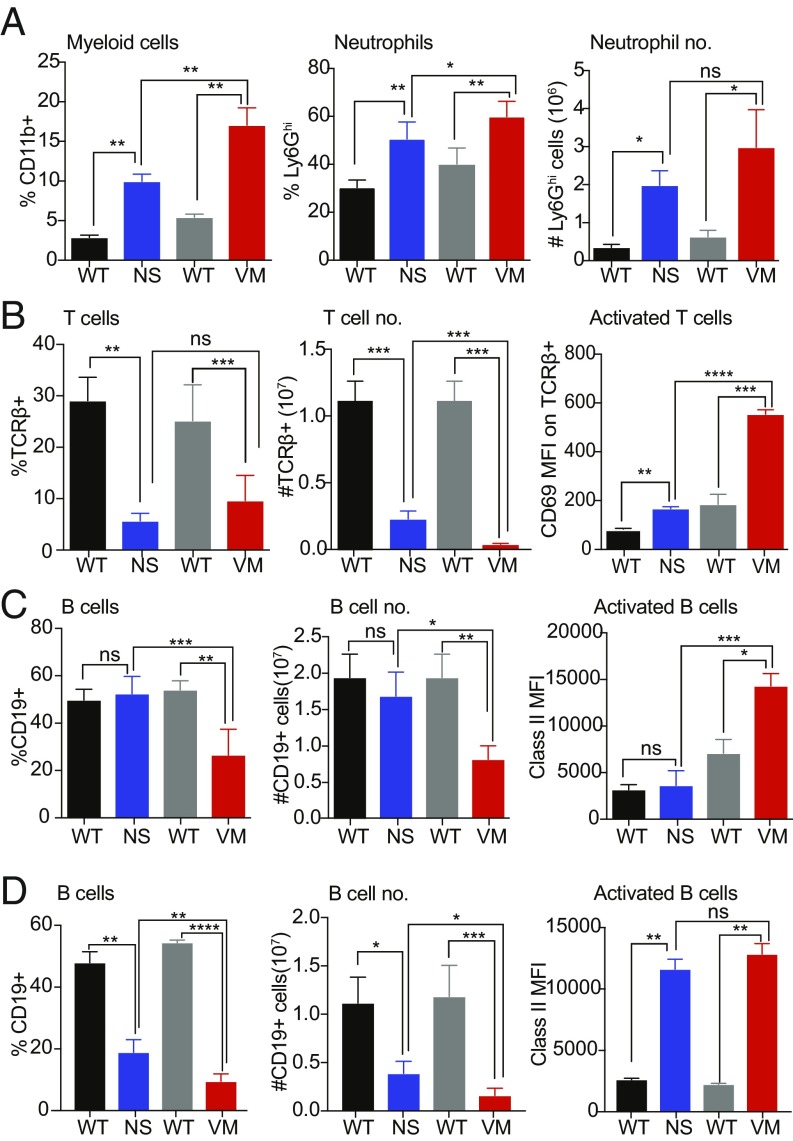
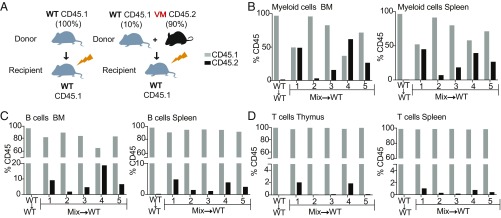
Studies over the past decade have revealed a central role for innate immune sensors in autoimmune and autoinflammatory diseases. cGAS, a cytosolic DNA sensor, detects both foreign and host DNA and generates a second-messenger cGAMP, which in turn binds and activates stimulator of IFN genes (STING), leading to induction of type I interferons and inflammatory cytokines. Recently, gain-of-function mutations in STING have been identified in patients with STING-associated vasculopathy with onset in infancy (SAVI). SAVI patients present with early-onset systemic inflammation and interstitial lung disease, resulting in pulmonary fibrosis and respiratory failure. Here, we describe two independent SAVI mouse models, harboring the two most common mutations found in patients. A direct comparison of these strains reveals a hierarchy of immune abnormalities, lung inflammation and fibrosis, which do not depend on either IFN-α/β receptor signaling or mixed lineage kinase domain-like pseudokinase (MLKL)-dependent necroptotic cell death pathways. Furthermore, radiation chimera experiments reveal how bone marrow from the V154M mutant mice transfer disease to the WT host, whereas the N153S does not, indicating mutation-specific disease outcomes. Moreover, using radiation chimeras we find that T cell lymphopenia depends on T cell-intrinsic expression of the SAVI mutation. Collectively, these mutant mice recapitulate many of the disease features seen in SAVI patients and highlight mutation-specific functions of STING that shed light on the heterogeneity observed in SAVI patients.
Keywords: SAVI; STING; T cells; cell death; type I interferonopathies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17:1142–1149. - PubMed
-
- Ahn J, Barber GN. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr Opin Immunol. 2014;31:121–126. - PubMed
-
- Crow YJ, Manel N. Aicardi-Goutières syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
