

Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia
- PMID: 30944321
- PMCID: PMC6447538
- DOI: 10.1038/s41467-019-09469-3
Proteogenomics and Hi-C reveal transcriptional dysregulation in high hyperdiploid childhood acute lymphoblastic leukemia
Abstract
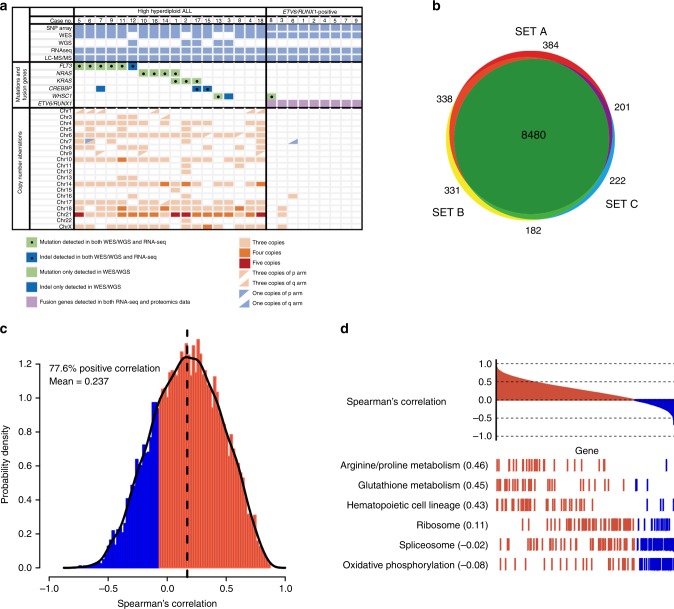
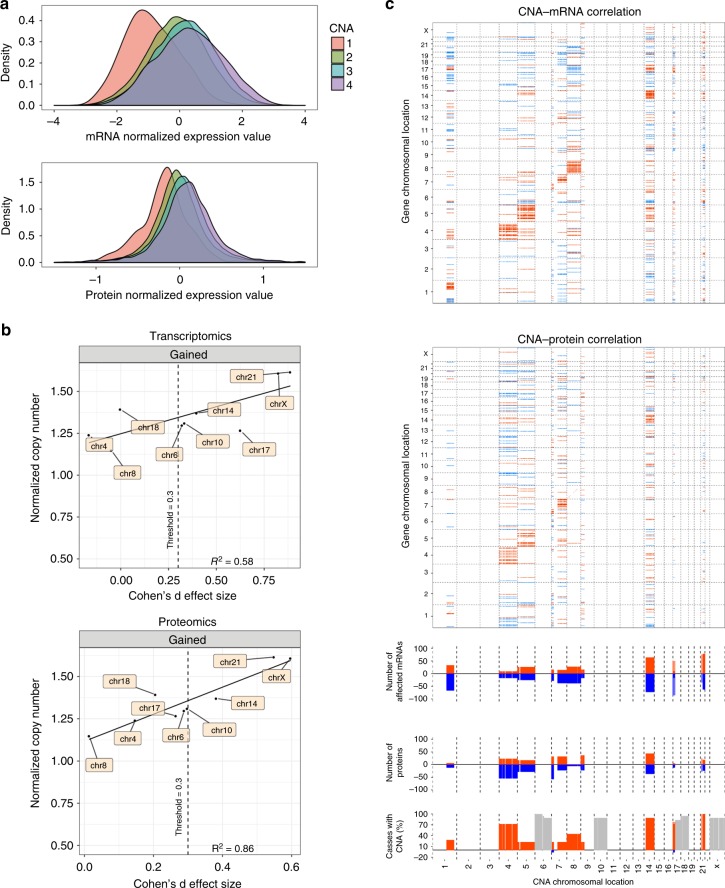
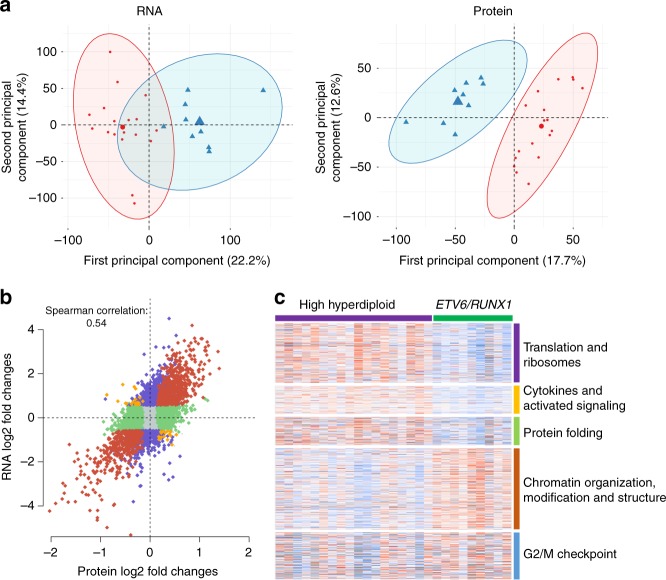
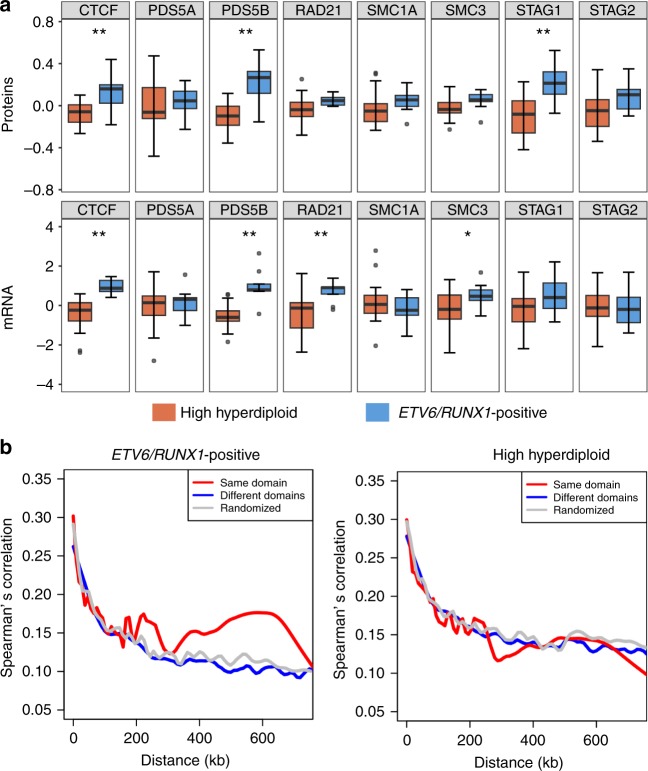
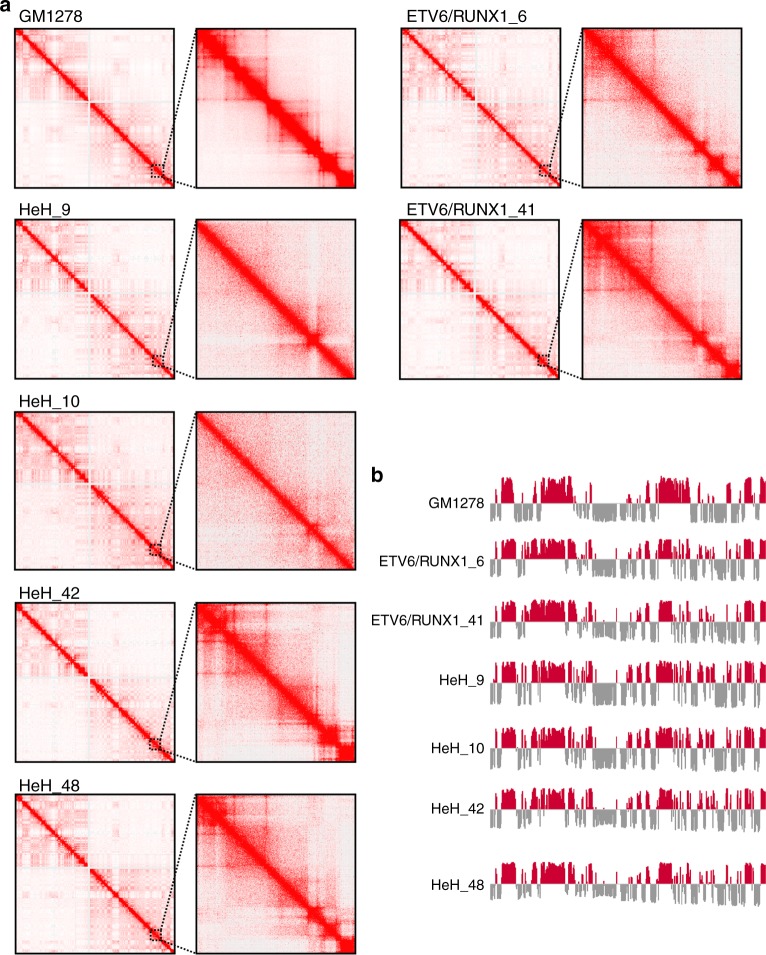
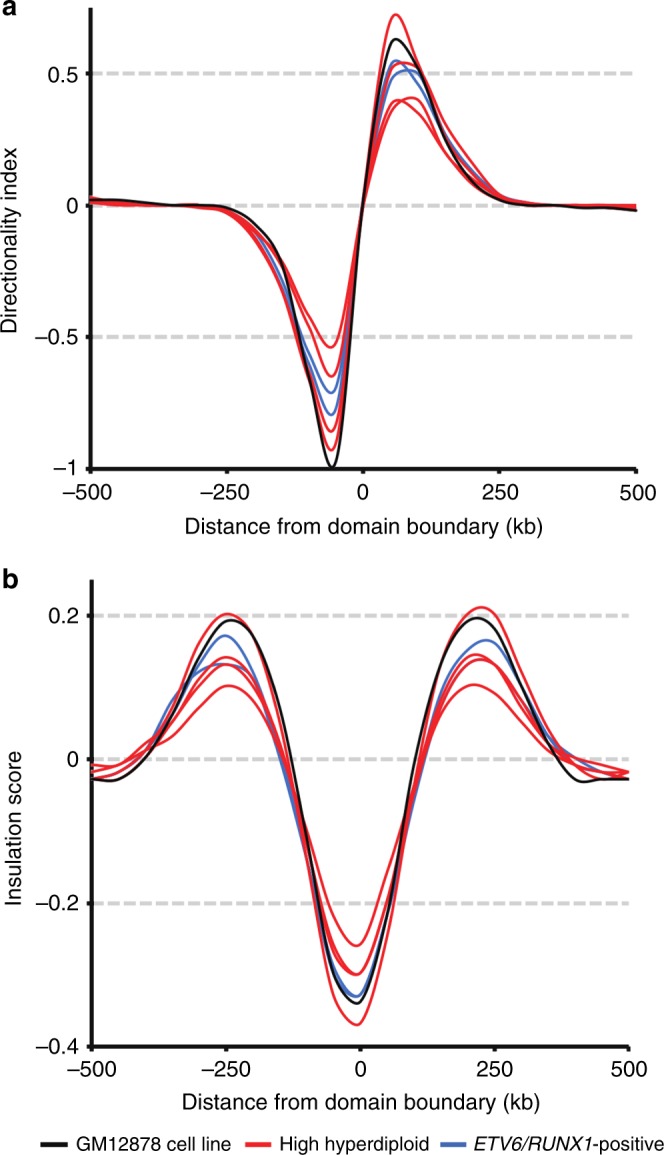
Hyperdiploidy, i.e. gain of whole chromosomes, is one of the most common genetic features of childhood acute lymphoblastic leukemia (ALL), but its pathogenetic impact is poorly understood. Here, we report a proteogenomic analysis on matched datasets from genomic profiling, RNA-sequencing, and mass spectrometry-based analysis of >8,000 genes and proteins as well as Hi-C of primary patient samples from hyperdiploid and ETV6/RUNX1-positive pediatric ALL. We show that CTCF and cohesin, which are master regulators of chromatin architecture, display low expression in hyperdiploid ALL. In line with this, a general genome-wide dysregulation of gene expression in relation to topologically associating domain (TAD) borders were seen in the hyperdiploid group. Furthermore, Hi-C of a limited number of hyperdiploid childhood ALL cases revealed that 2/4 cases displayed a clear loss of TAD boundary strength and 3/4 showed reduced insulation at TAD borders, with putative leukemogenic effects.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
