

Genetic compensation triggered by mutant mRNA degradation
- PMID: 30944477
- PMCID: PMC6707827
- DOI: 10.1038/s41586-019-1064-z
Genetic compensation triggered by mutant mRNA degradation
Abstract
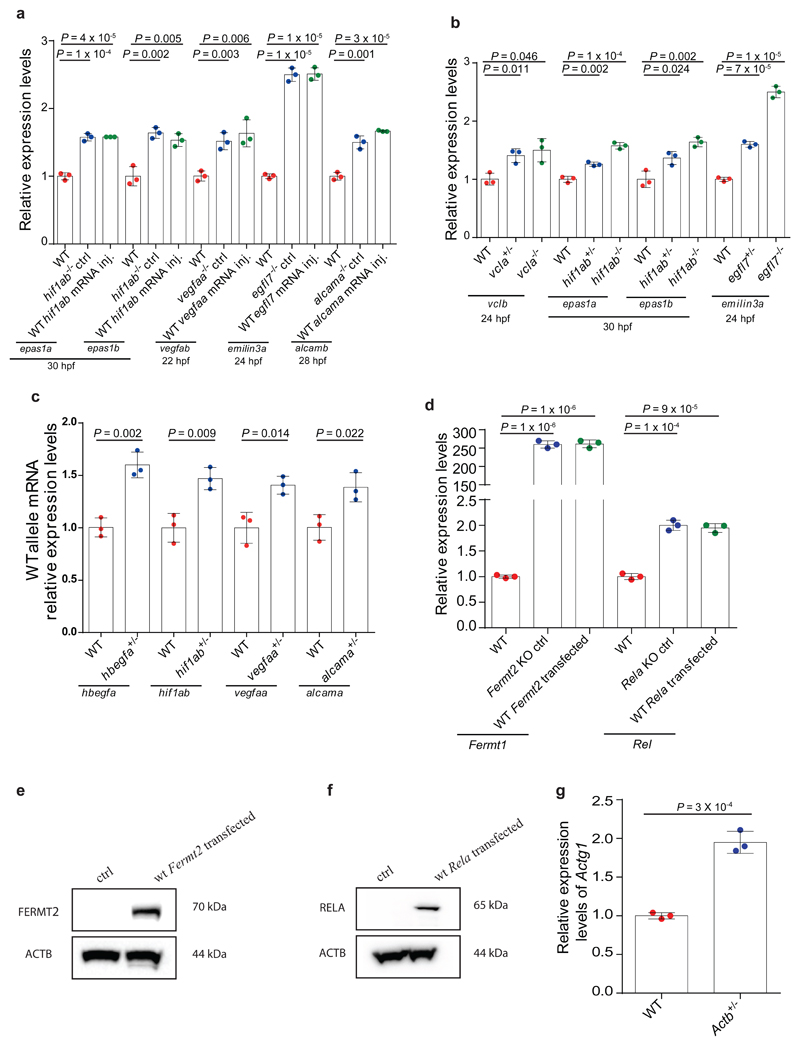
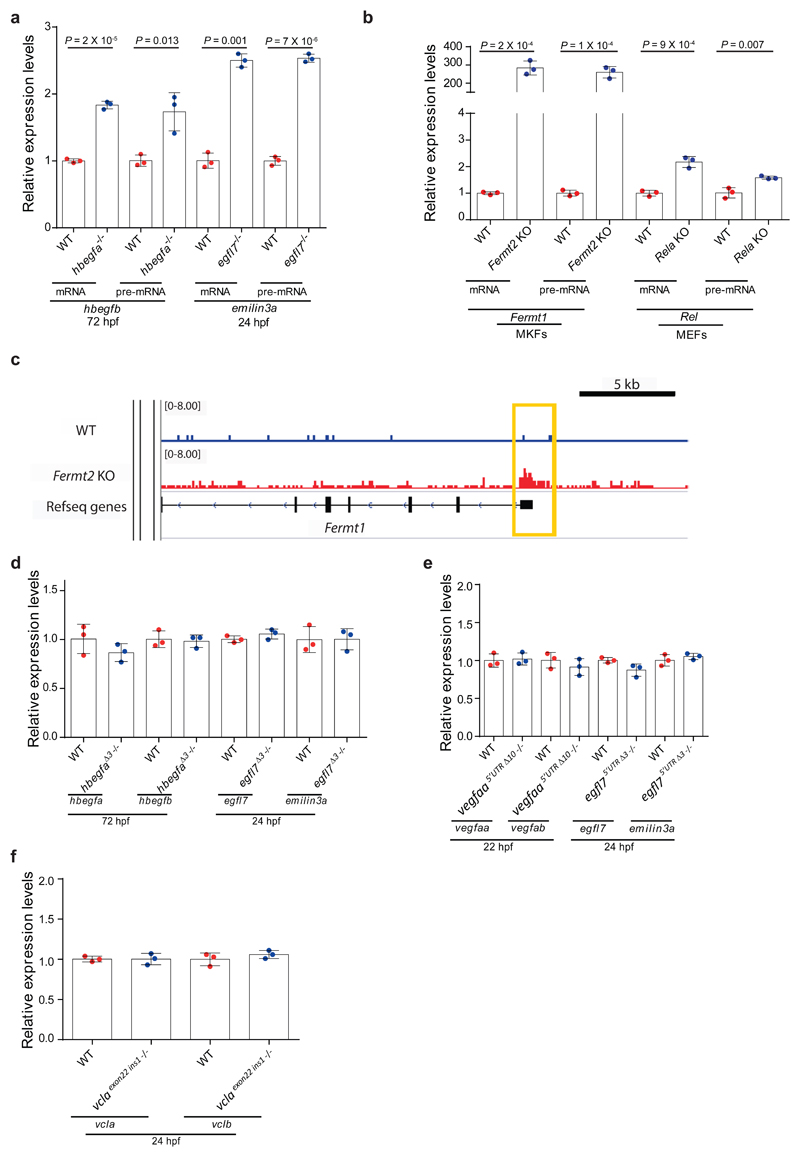
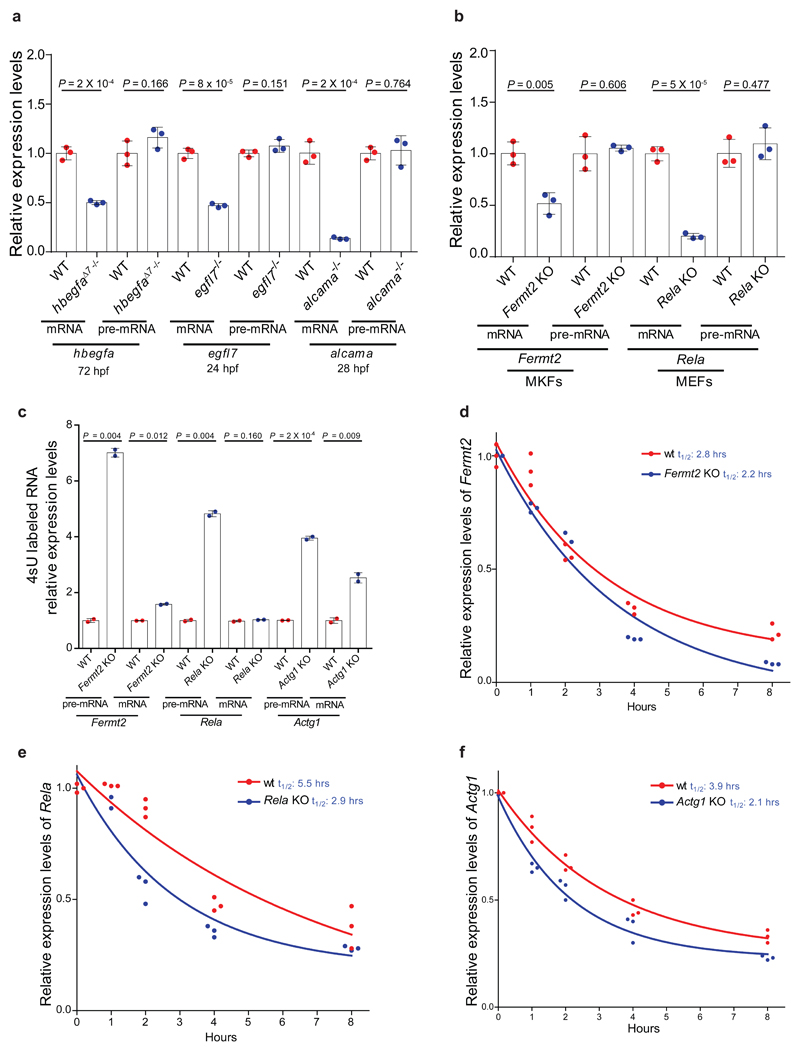
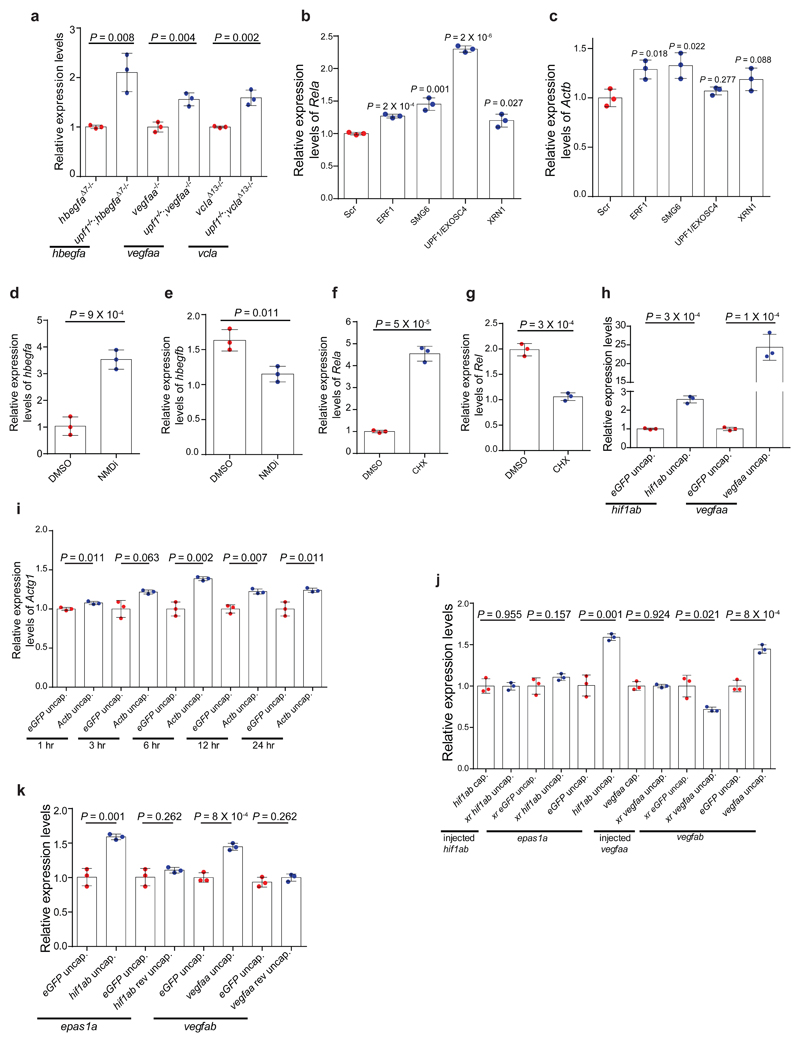
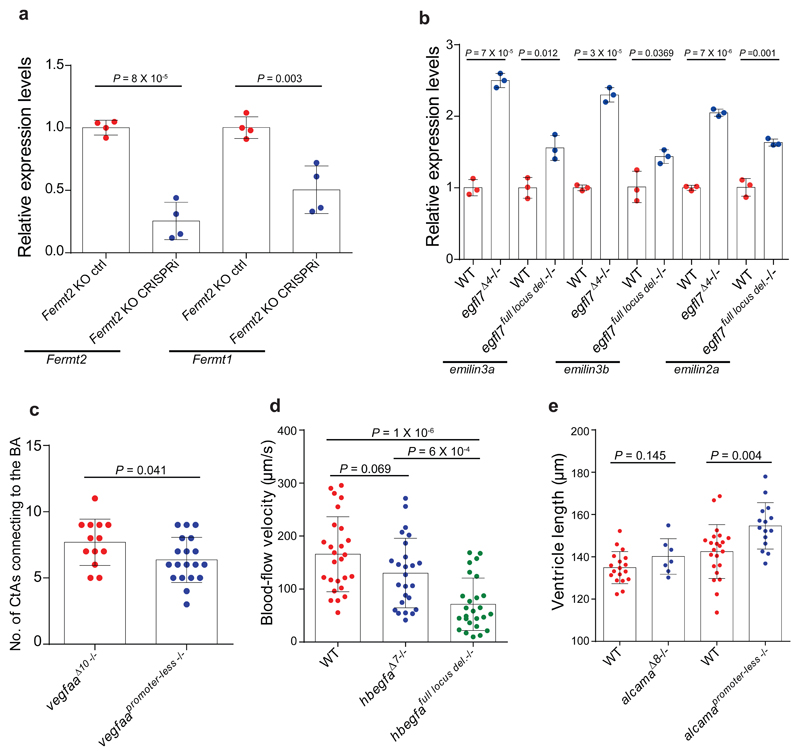
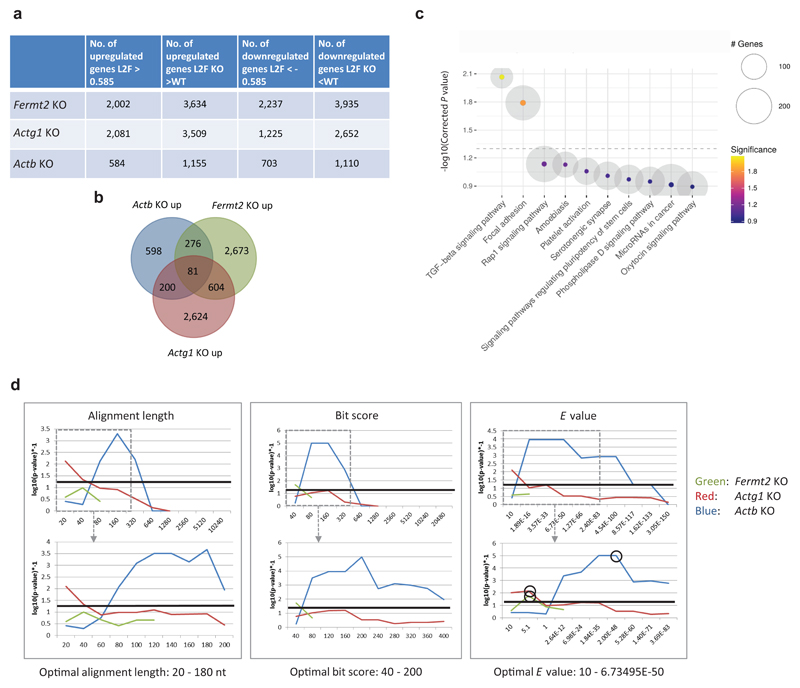
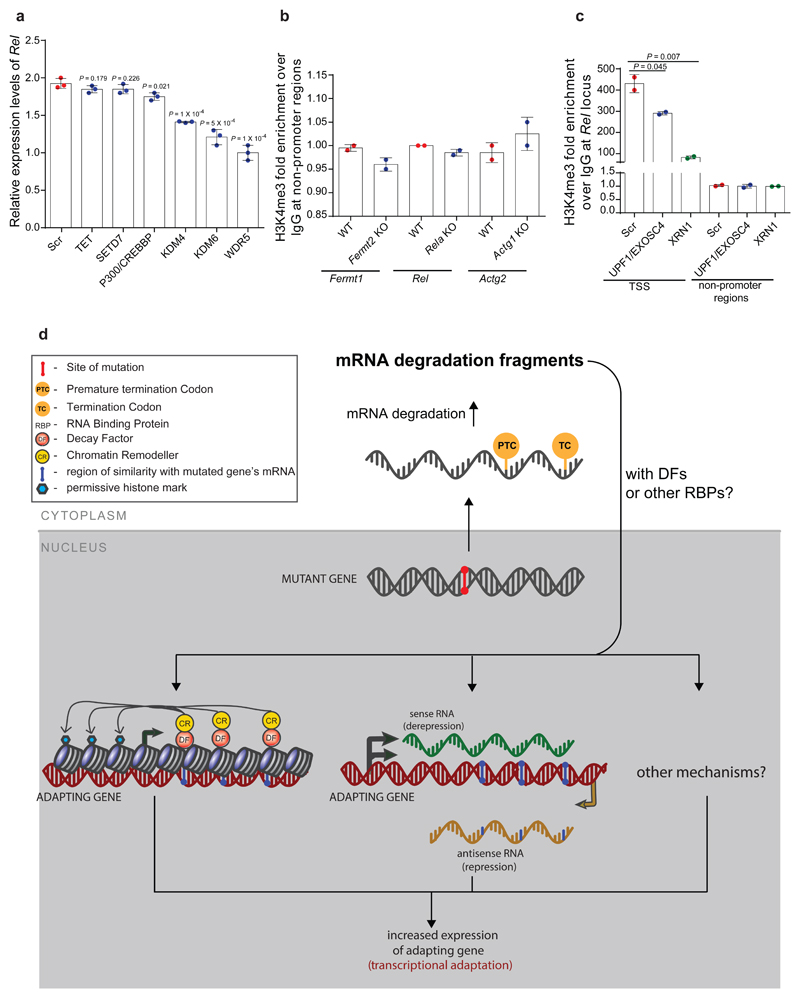
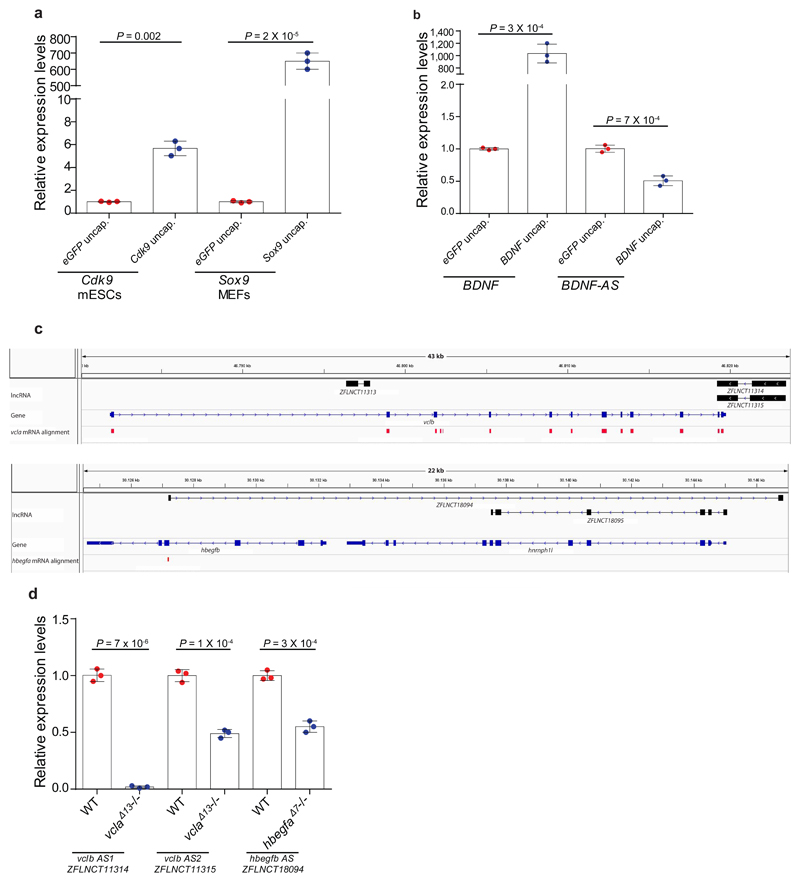
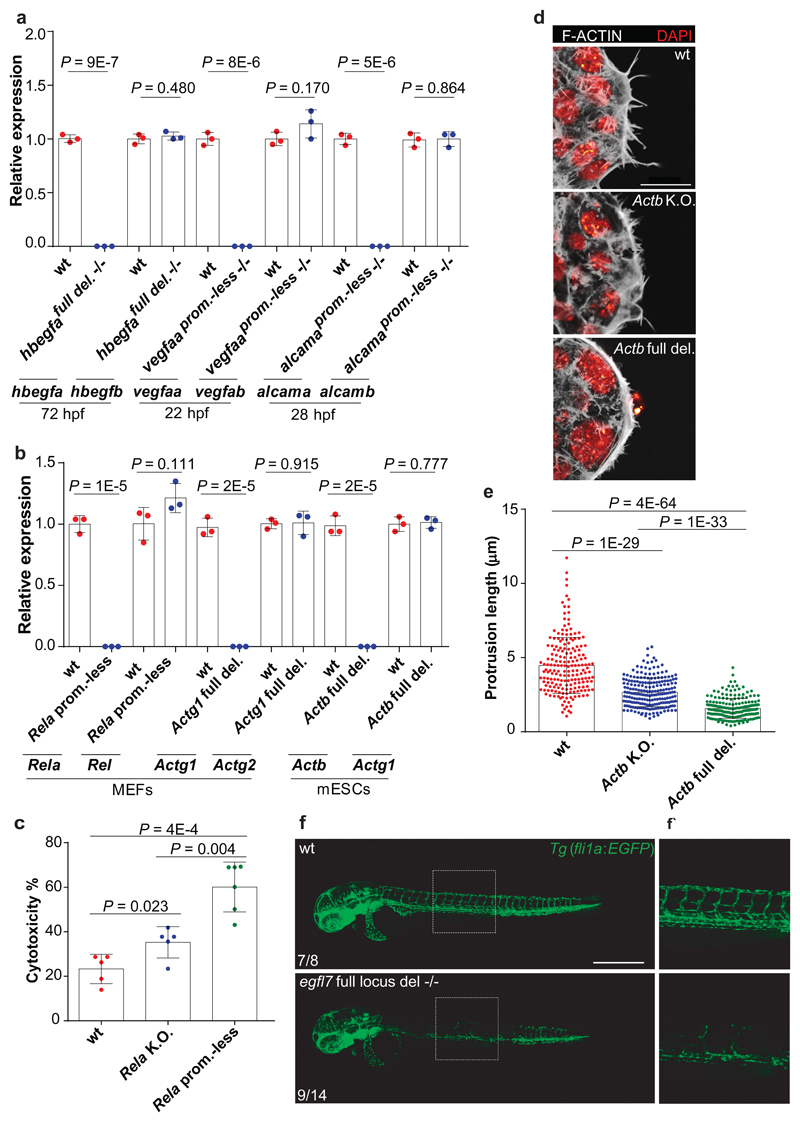
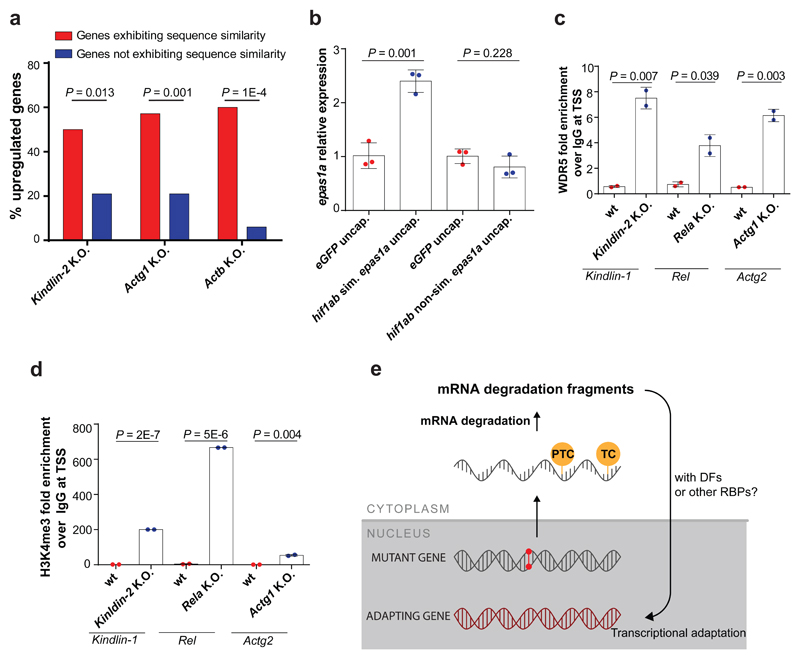
Genetic robustness, or the ability of an organism to maintain fitness in the presence of harmful mutations, can be achieved via protein feedback loops. Previous work has suggested that organisms may also respond to mutations by transcriptional adaptation, a process by which related gene(s) are upregulated independently of protein feedback loops. However, the prevalence of transcriptional adaptation and its underlying molecular mechanisms are unknown. Here, by analysing several models of transcriptional adaptation in zebrafish and mouse, we uncover a requirement for mutant mRNA degradation. Alleles that fail to transcribe the mutated gene do not exhibit transcriptional adaptation, and these alleles give rise to more severe phenotypes than alleles displaying mutant mRNA decay. Transcriptome analysis in alleles displaying mutant mRNA decay reveals the upregulation of a substantial proportion of the genes that exhibit sequence similarity with the mutated gene's mRNA, suggesting a sequence-dependent mechanism. These findings have implications for our understanding of disease-causing mutations, and will help in the design of mutant alleles with minimal transcriptional adaptation-derived compensation.
Conflict of interest statement
The authors declare no competing interests.
Figures




Comment in
-
Genetic paradox explained by nonsense.Nature. 2019 Apr;568(7751):179-180. doi: 10.1038/d41586-019-00823-5. Nature. 2019. PMID: 30962551 Free PMC article.
-
When all is not lost: considering genetic compensation in laboratory animals.Lab Anim (NY). 2019 Oct;48(10):282-284. doi: 10.1038/s41684-019-0397-4. Lab Anim (NY). 2019. PMID: 31477900 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
