

Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory
- PMID: 30948719
- PMCID: PMC6449405
- DOI: 10.1038/s41467-019-09483-5
Dynamics of genome reorganization during human cardiogenesis reveal an RBM20-dependent splicing factory
Abstract
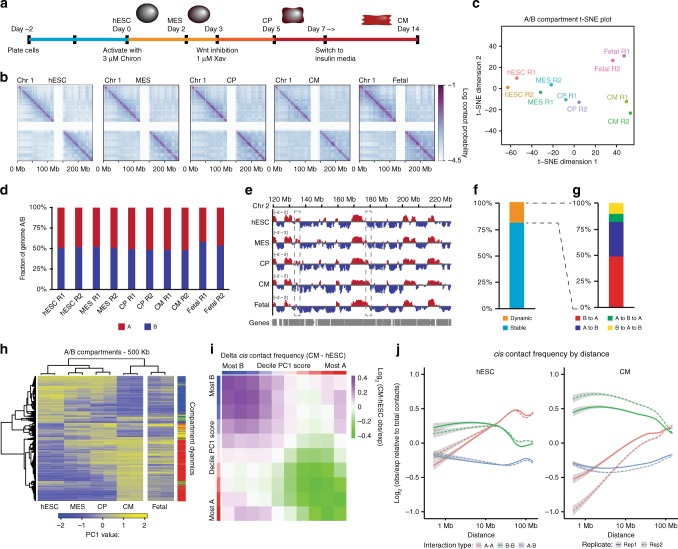
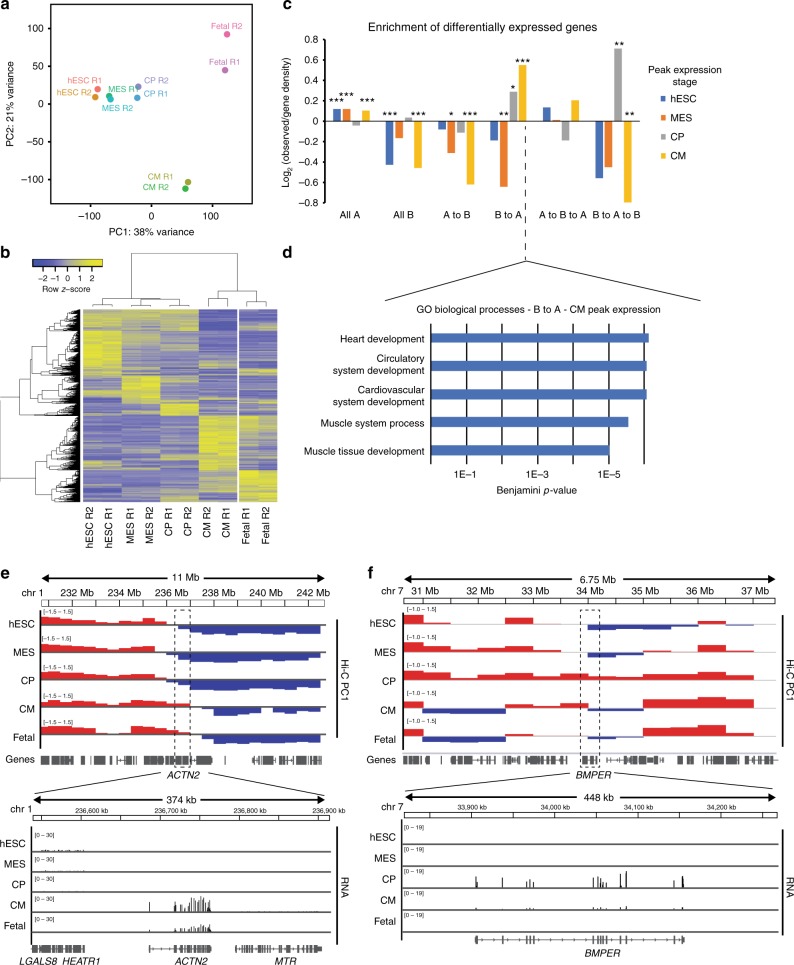
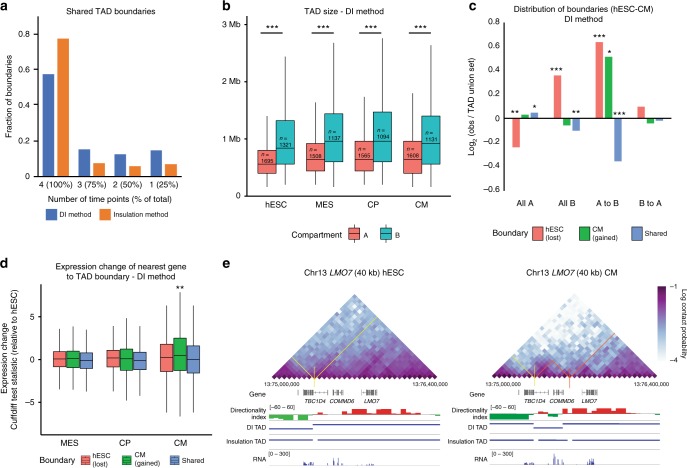
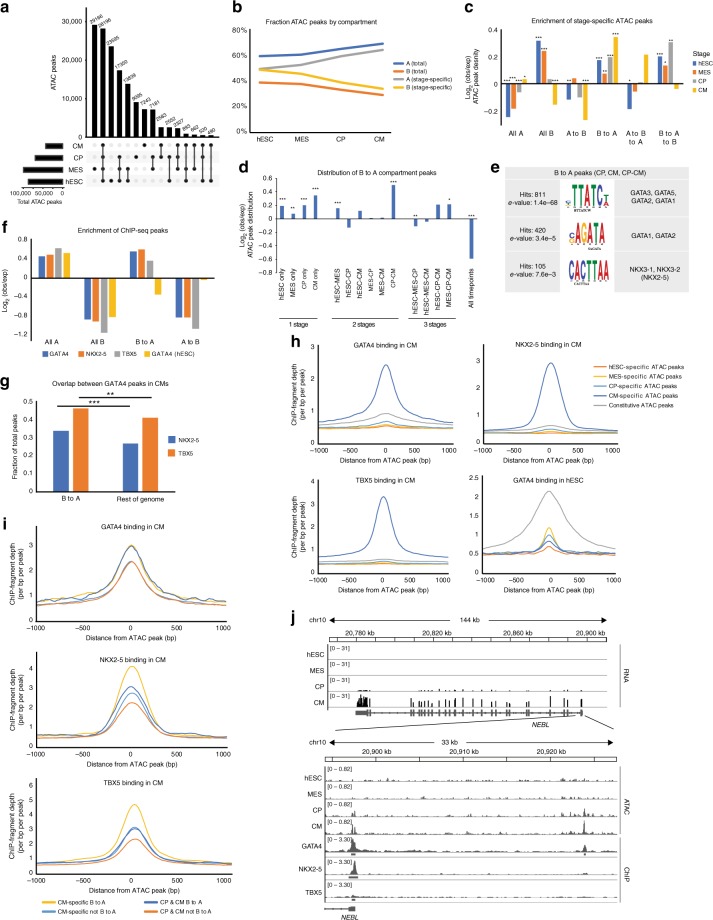
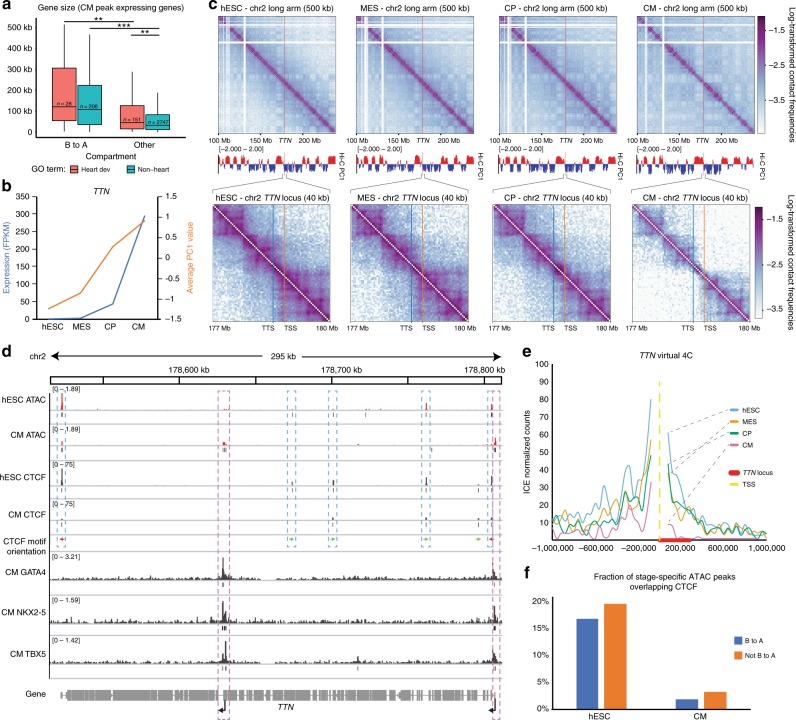
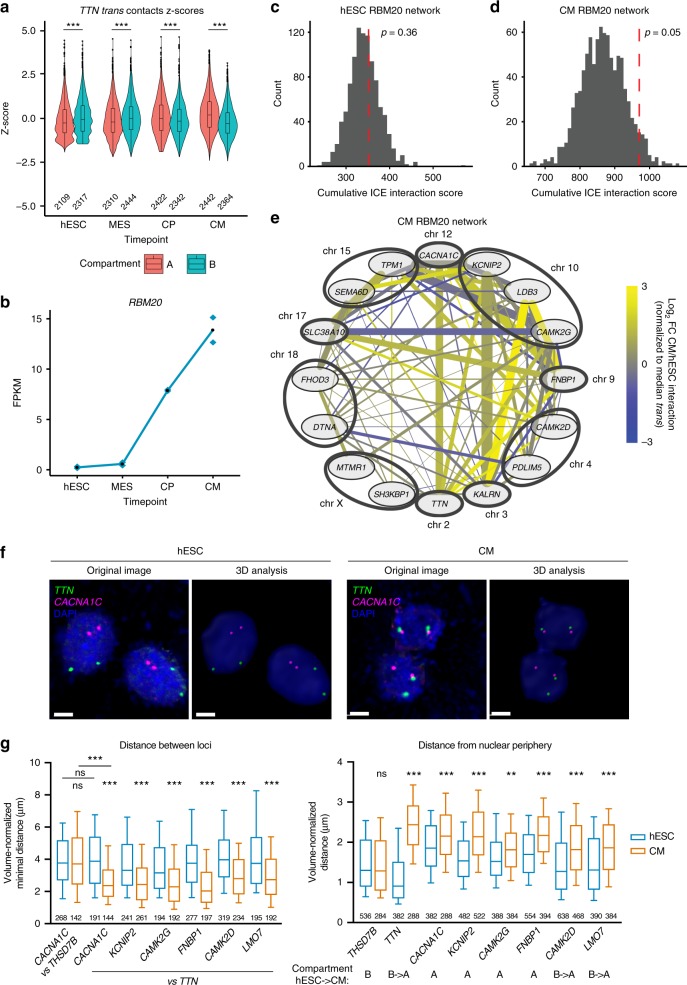
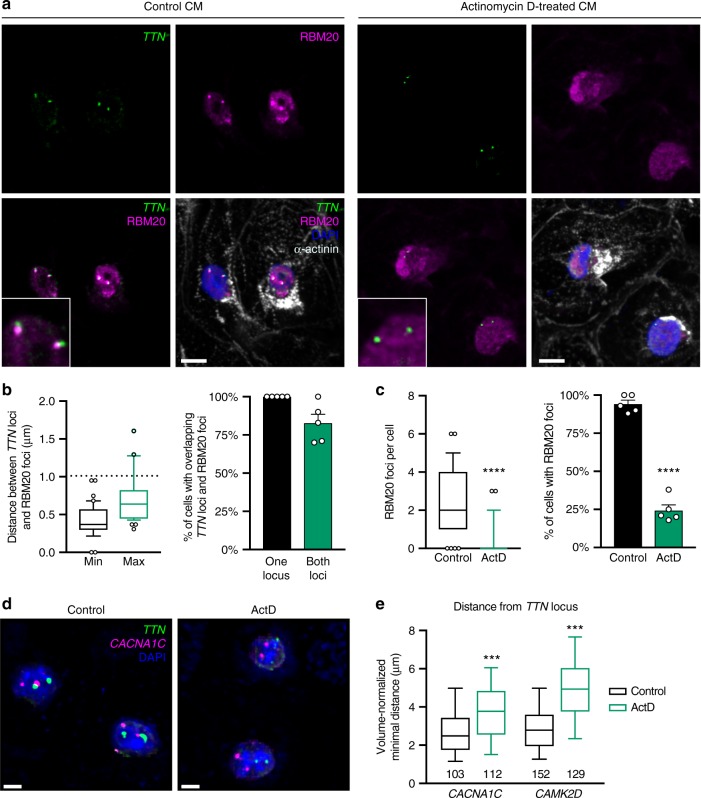
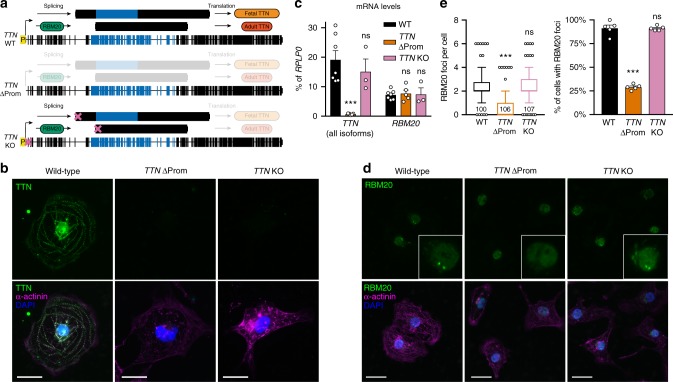
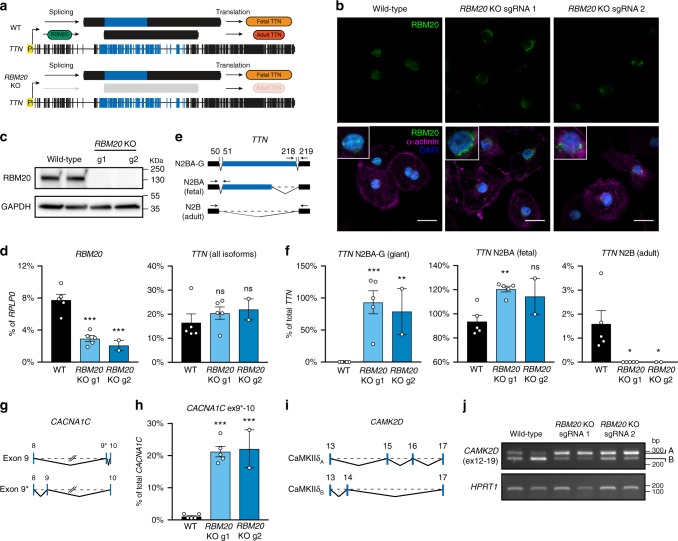
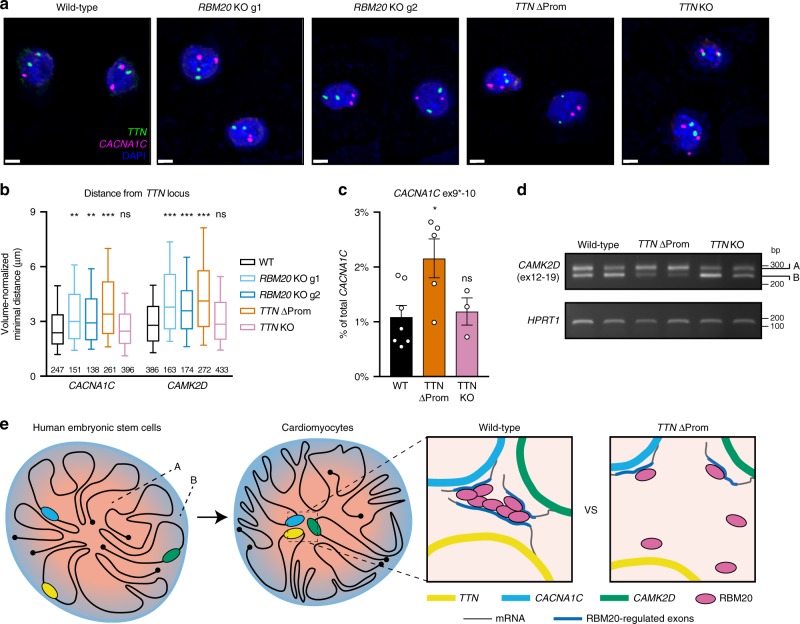
Functional changes in spatial genome organization during human development are poorly understood. Here we report a comprehensive profile of nuclear dynamics during human cardiogenesis from pluripotent stem cells by integrating Hi-C, RNA-seq and ATAC-seq. While chromatin accessibility and gene expression show complex on/off dynamics, large-scale genome architecture changes are mostly unidirectional. Many large cardiac genes transition from a repressive to an active compartment during differentiation, coincident with upregulation. We identify a network of such gene loci that increase their association inter-chromosomally, and are targets of the muscle-specific splicing factor RBM20. Genome editing studies show that TTN pre-mRNA, the main RBM20-regulated transcript in the heart, nucleates RBM20 foci that drive spatial proximity between the TTN locus and other inter-chromosomal RBM20 targets such as CACNA1C and CAMK2D. This mechanism promotes RBM20-dependent alternative splicing of the resulting transcripts, indicating the existence of a cardiac-specific trans-interacting chromatin domain (TID) functioning as a splicing factory.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
