

Within-Species Genomic Variation and Variable Patterns of Recombination in the Tetracycline Producer Streptomyces rimosus
- PMID: 30949149
- PMCID: PMC6437091
- DOI: 10.3389/fmicb.2019.00552
Within-Species Genomic Variation and Variable Patterns of Recombination in the Tetracycline Producer Streptomyces rimosus
Abstract
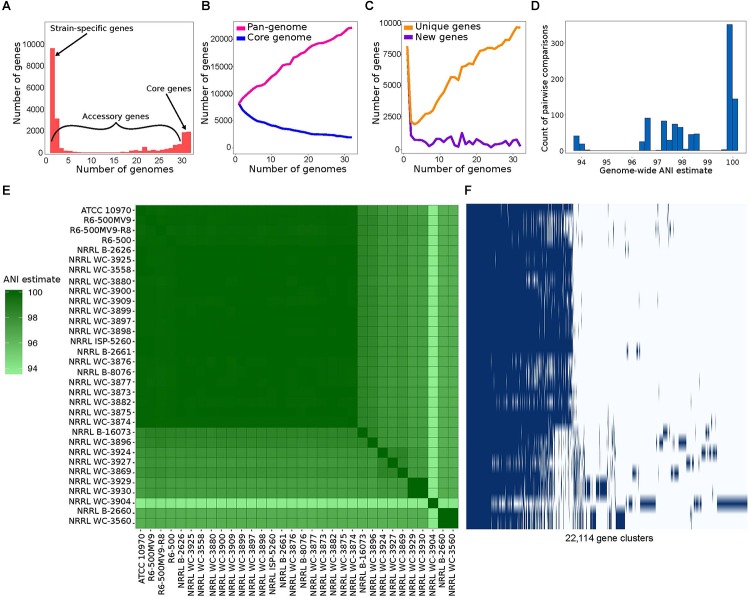
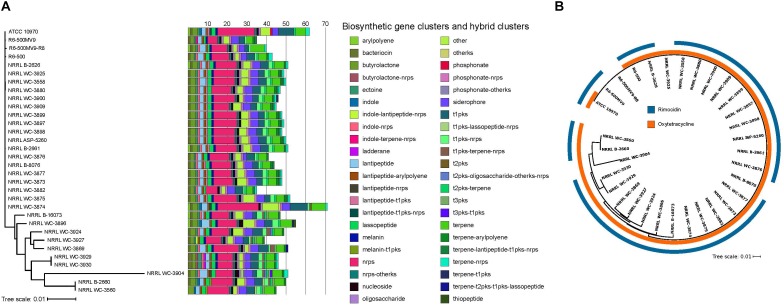
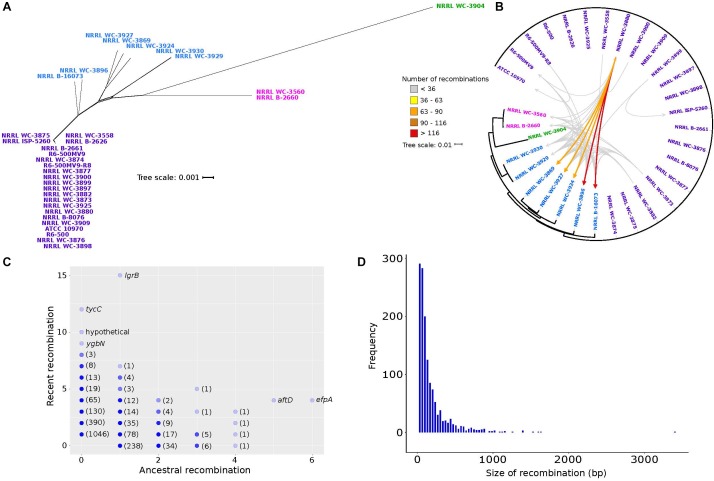
Streptomyces rimosus is best known as the primary source of the tetracycline class of antibiotics, most notably oxytetracycline, which have been widely used against many gram-positive and gram-negative pathogens and protozoan parasites. However, despite the medical and agricultural importance of S. rimosus, little is known of its evolutionary history and genome dynamics. In this study, we aim to elucidate the pan-genome characteristics and phylogenetic relationships of 32 S. rimosus genomes. The S. rimosus pan-genome contains more than 22,000 orthologous gene clusters, and approximately 8.8% of these genes constitutes the core genome. A large part of the accessory genome is composed of 9,646 strain-specific genes. S. rimosus exhibits an open pan-genome (decay parameter α = 0.83) and high gene diversity between strains (genomic fluidity φ = 0.12). We also observed strain-level variation in the distribution and abundance of biosynthetic gene clusters (BGCs) and that each individual S. rimosus genome has a unique repertoire of BGCs. Lastly, we observed variation in recombination, with some strains donating or receiving DNA more often than others, strains that tend to frequently recombine with specific partners, genes that often experience recombination more than others, and variable sizes of recombined DNA sequences. We conclude that the high levels of inter-strain genomic variation in S. rimosus is partly explained by differences in recombination among strains. These results have important implications on current efforts for natural drug discovery, the ecological role of strain-level variation in microbial populations, and addressing the fundamental question of why microbes have pan-genomes.
Keywords: Streptomyces; accessory genome; biosynthetic gene cluster; core genome; pan-genome; recombination; tetracycline.
Figures
References
-
- Andam C. P., Mitchell P. K., Callendrello A., Chang Q., Corander J., Chaguza C., et al. (2017). Genomic epidemiology of penicillin-nonsusceptible pneumococci with nonvaccine serotypes causing invasive disease in the United States. J. Clin. Microbiol. 55 1104–1115. 10.1128/JCM.02453-16 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources