

Leveraging Spatial Variation in Tumor Purity for Improved Somatic Variant Calling of Archival Tumor Only Samples
- PMID: 30949446
- PMCID: PMC6435595
- DOI: 10.3389/fonc.2019.00119
Leveraging Spatial Variation in Tumor Purity for Improved Somatic Variant Calling of Archival Tumor Only Samples
Abstract
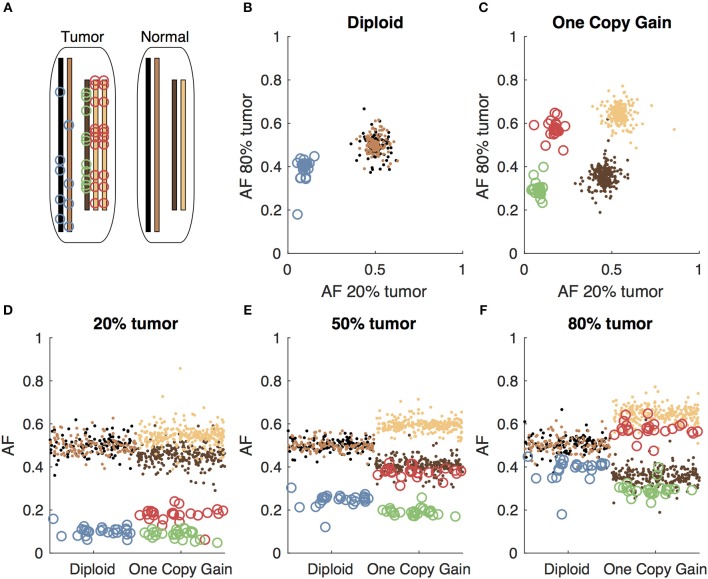
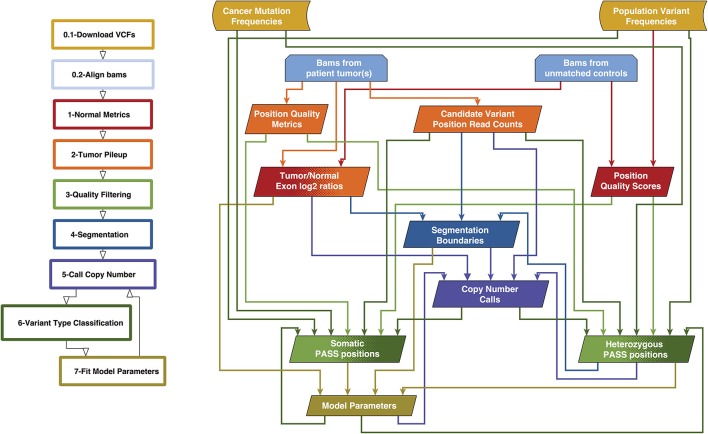
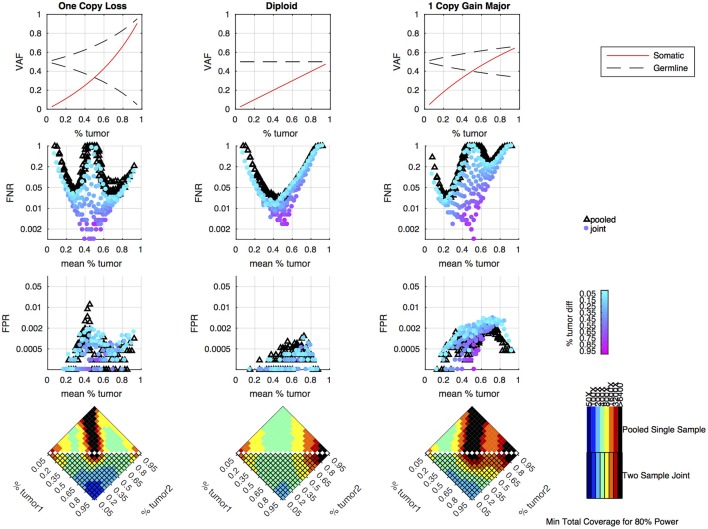
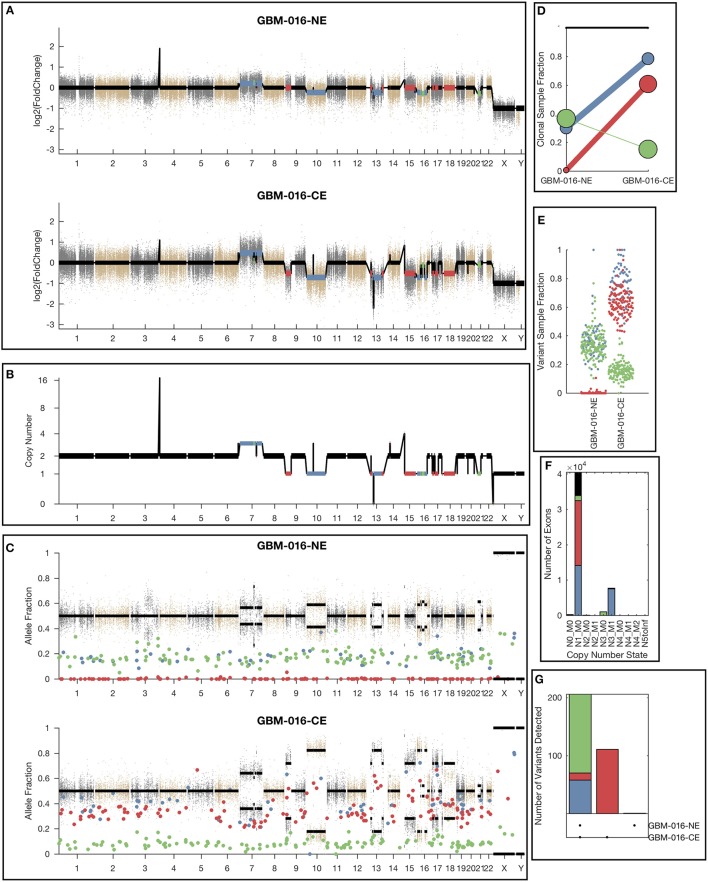
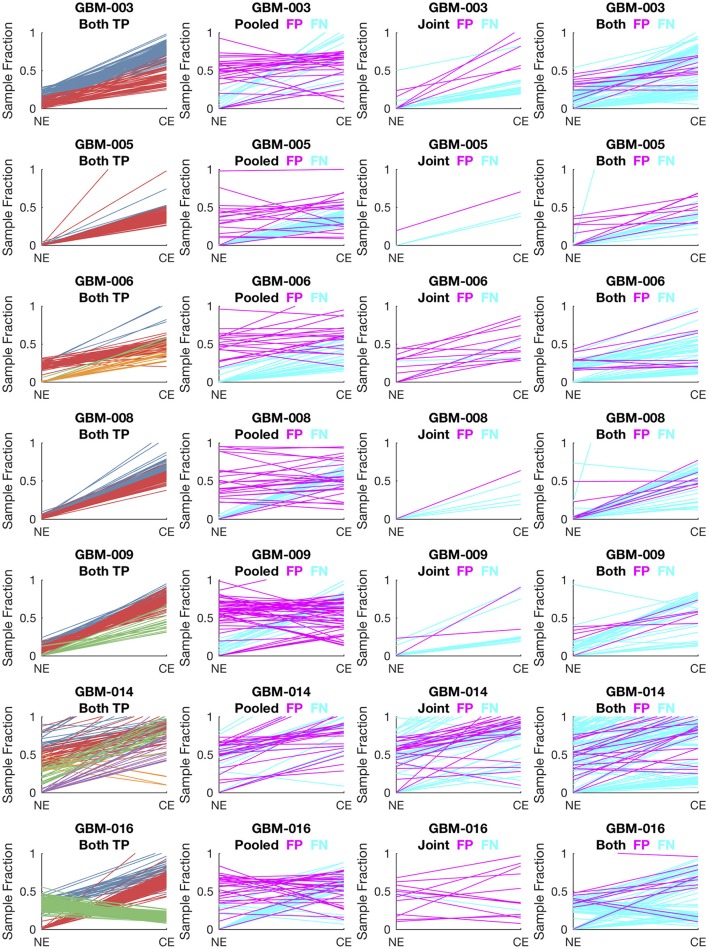
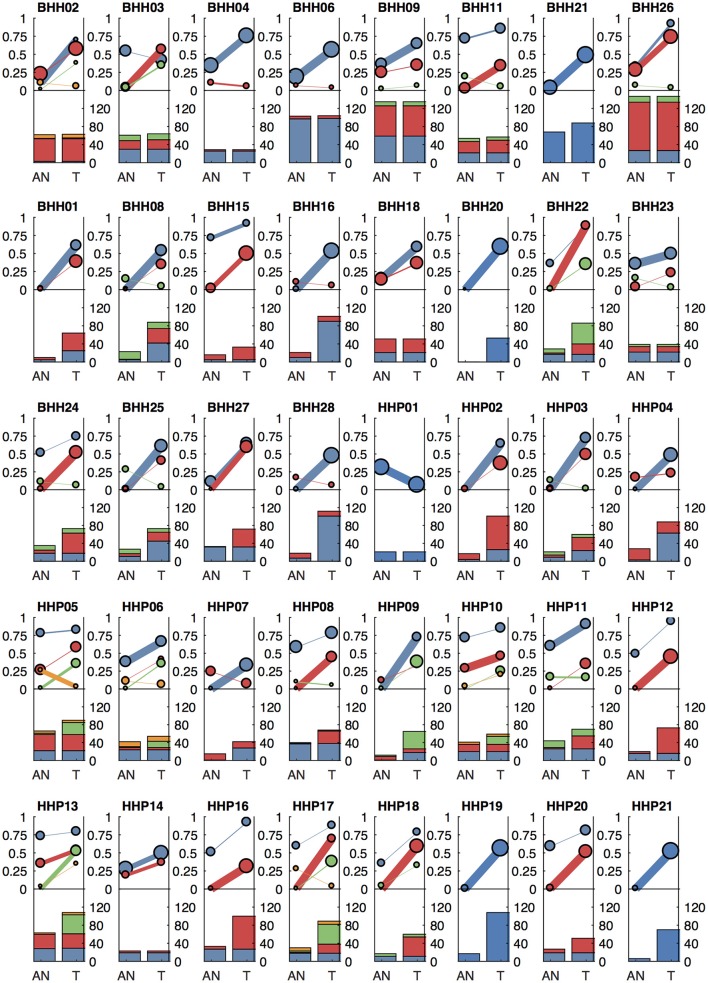
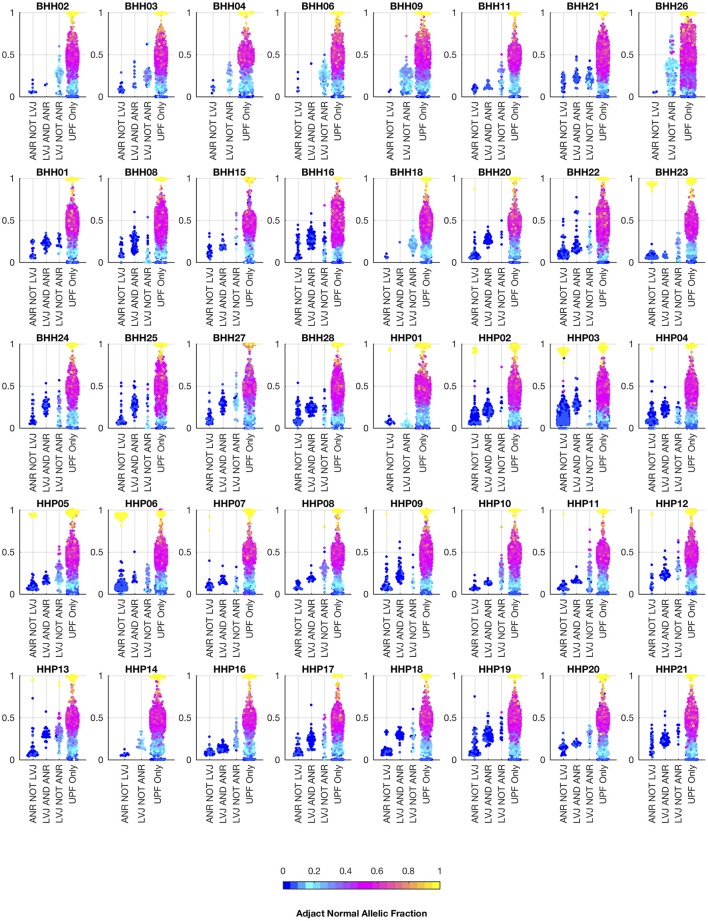
Archival tumor samples represent a rich resource of annotated specimens for translational genomics research. However, standard variant calling approaches require a matched normal sample from the same individual, which is often not available in the retrospective setting, making it difficult to distinguish between true somatic variants and individual-specific germline variants. Archival sections often contain adjacent normal tissue, but this tissue can include infiltrating tumor cells. As existing comparative somatic variant callers are designed to exclude variants present in the normal sample, a novel approach is required to leverage adjacent normal tissue with infiltrating tumor cells for somatic variant calling. Here we present lumosVar 2.0, a software package designed to jointly analyze multiple samples from the same patient, built upon our previous single sample tumor only variant caller lumosVar 1.0. The approach assumes that the allelic fraction of somatic variants and germline variants follow different patterns as tumor content and copy number state change. lumosVar 2.0 estimates allele specific copy number and tumor sample fractions from the data, and uses a to model to determine expected allelic fractions for somatic and germline variants and to classify variants accordingly. To evaluate the utility of lumosVar 2.0 to jointly call somatic variants with tumor and adjacent normal samples, we used a glioblastoma dataset with matched high and low tumor content and germline whole exome sequencing data (for true somatic variants) available for each patient. Both sensitivity and positive predictive value were improved when analyzing the high tumor and low tumor samples jointly compared to analyzing the samples individually or in-silico pooling of the two samples. Finally, we applied this approach to a set of breast and prostate archival tumor samples for which tumor blocks containing adjacent normal tissue were available for sequencing. Joint analysis using lumosVar 2.0 detected several variants, including known cancer hotspot mutations that were not detected by standard somatic variant calling tools using the adjacent tissue as presumed normal reference. Together, these results demonstrate the utility of leveraging paired tissue samples to improve somatic variant calling when a constitutional sample is not available.
Keywords: cancer genomics; cancer hotspot mutations; next generation sequencing; somatic variant calling; tumor exome sequencing; tumor-only sequencing.
Figures
References
-
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. . Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. (2015) 17:251–64. 10.1016/j.jmoldx.2014.12.006 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous