

Lysosomal Pathways and Autophagy Distinctively Control Endothelial Cell Behavior to Affect Tumor Vasculature
- PMID: 30949450
- PMCID: PMC6435524
- DOI: 10.3389/fonc.2019.00171
Lysosomal Pathways and Autophagy Distinctively Control Endothelial Cell Behavior to Affect Tumor Vasculature
Abstract
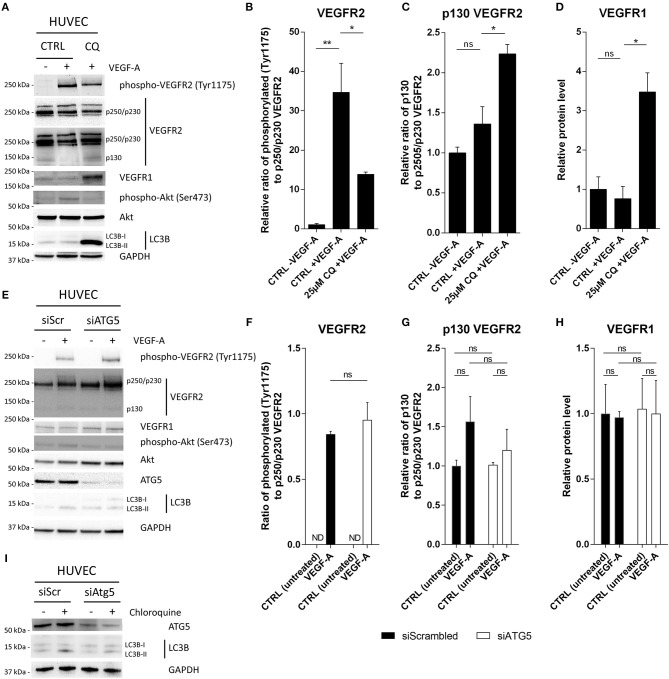
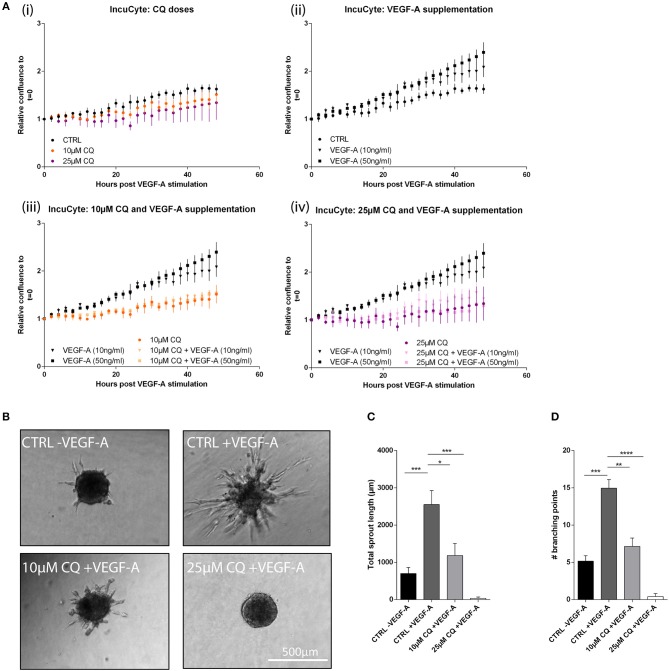
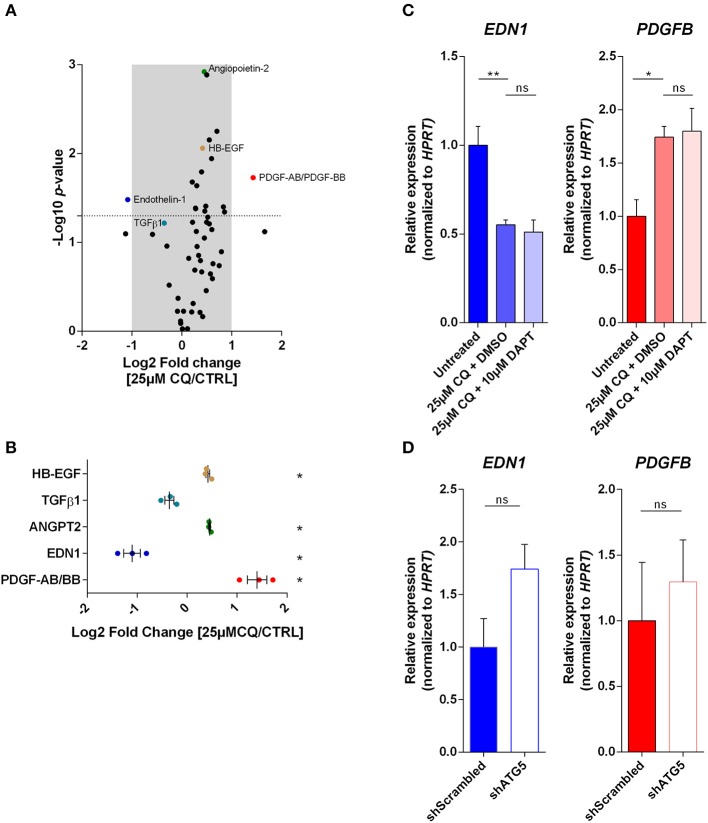
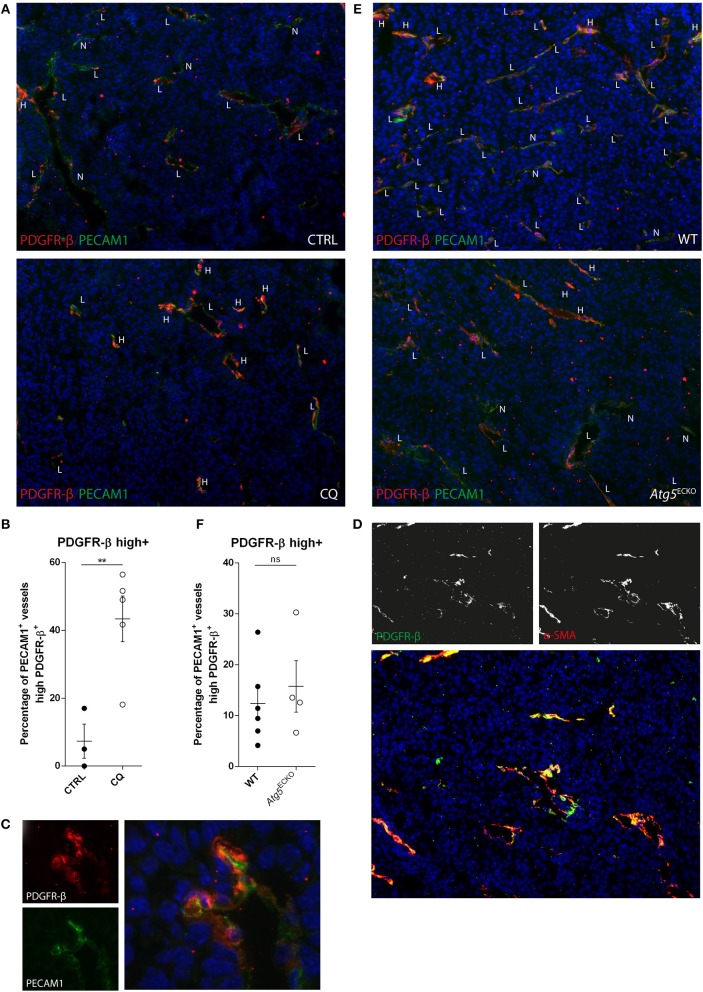
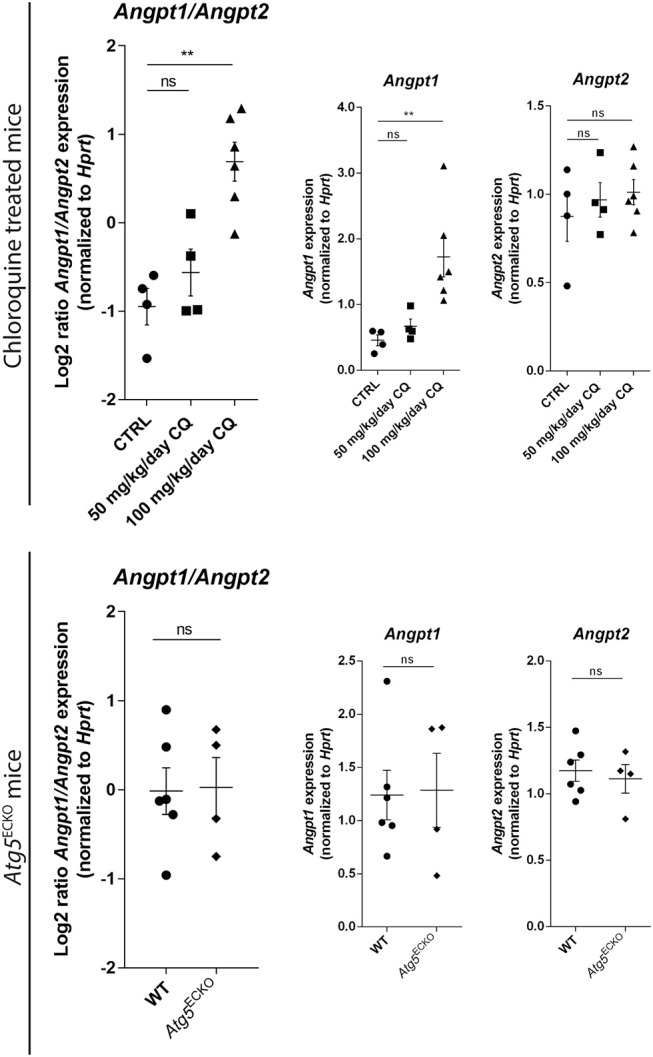
Cancer cell-stromal cell crosstalk is orchestrated by a plethora of ligand-receptor interactions generating a tumor microenvironment (TME) which favors tumor growth. The high pro-angiogenic nature of the TME perpetuates the chaotic network of structurally immature, low pericyte-covered vessels characteristic of the tumor vasculature. We previously demonstrated that chloroquine (CQ) -a lysosomotropic agent used as first-generation autophagy blocker in clinical trials- induced tumor vessel normalization and reduced tumor hypoxia. CQ improved both vessel structure and maturation, whereas the conditional knockout of the crucial autophagy gene Atg5 in endothelial cells (ECs) did not, thus highlighting a potential differential role for EC-associated autophagy and the lysosomes in pathological tumor angiogenesis. However, how CQ or ATG5-deficiency in ECs affect angiogenic signals regulating EC-pericyte interface and therefore vessel maturation, remains unknown. Here, we show that in ECs CQ constrained VEGF-A-mediated VEGF receptor (VEGFR)2 phosphorylation, a driver of angiogenic signaling. In the presence of CQ we observed increased expression of the decoy receptor VEGFR1 and of a lower molecular weight form of VEGFR2, suggesting receptor cleavage. Consequently, VEGF-A-driven EC spheroid sprouting was reduced by CQ treatment. Furthermore, CQ significantly affected the transcription and secretion of platelet-derived growth factor (PDGF)-AB/BB (upregulated) and Endothelin-1 (EDN1, downregulated), both modulators of perivascular cell (PC) behavior. In contrast, silencing of ATG5 in ECs had no effect on VEGFR2 to VEGFR1 ratio nor on PDGFB and EDN1 expression. Accordingly, mice harboring B16F10 melanoma tumors treated with CQ, displayed both an increased number of αSMA+ PCs covering tumor vessels and co-expressed PDGF receptor-β, enabling PDGF ligand dependent recruitment. Moreover, upon CQ treatment the tumoral expression of angiopoietin-1 (Angpt1), which retains mural cells, and induces vessel stabilization by binding to the EC-localized cognate receptor (TIE2), was increased thus supporting the vessel normalization function of CQ. These features associated with improved tumor vasculature were not phenocopied by the specific deletion of Atg5 in ECs. In conclusion, this study further unravels endothelial cell autonomous and non-autonomous mechanisms by which CQ "normalizes" the intercellular communication in the tumor vasculature independent of autophagy.
Keywords: Angiopoietin1; Autophagy; PDGFR-β; VEGF/VEGFR-axis; intercellular crosstalk; tumor endothelial cells.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous