

Autosomal Dominant Leukodystrophy: A Disease of the Nuclear Lamina
- PMID: 30949481
- PMCID: PMC6435485
- DOI: 10.3389/fcell.2019.00041
Autosomal Dominant Leukodystrophy: A Disease of the Nuclear Lamina
Abstract
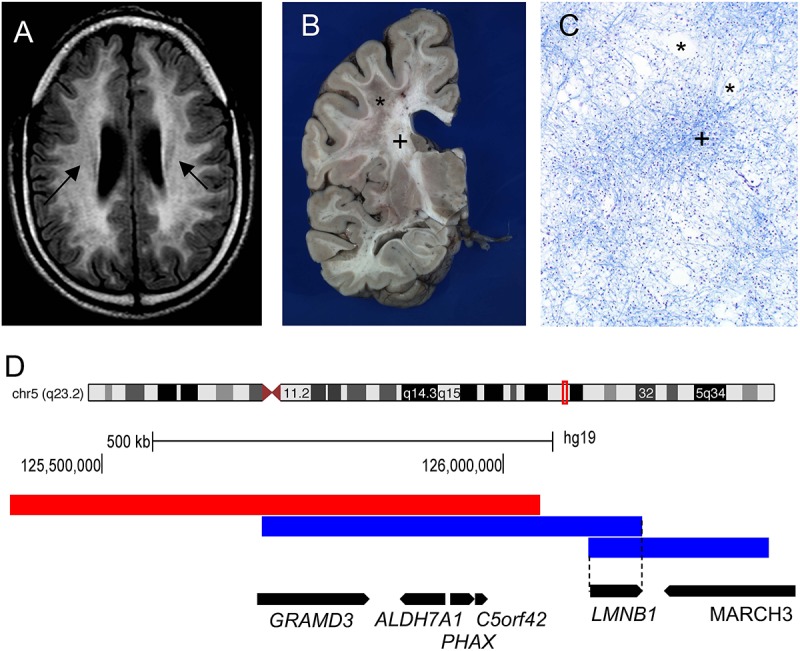
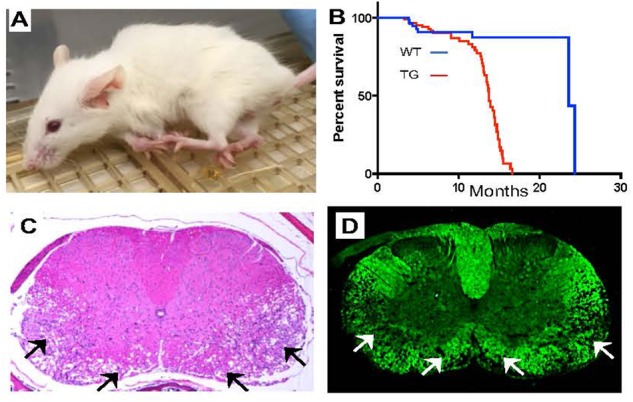
The nuclear lamina is a fibrous meshwork of proteins found adjacent to the inner nuclear membrane that plays a critical role in the maintenance of nuclear architecture. Made up of A and B type lamins, the nuclear lamina has recently been shown to contribute to numerous cellular functions such as chromatin organization, DNA replication, cellular proliferation, senescence, and aging. While at least a dozen disorders are associated with LMNA, the focus of this review is Autosomal Dominant Leukodystrophy (ADLD), the only disease associated with the lamin B1 gene (LMNB1). ADLD is a fatal, adult onset CNS demyelinating disorder that is caused by either genomic duplications involving LMNB1 or deletions upstream of the gene. Both mutation types result in increased LMNB1 gene expression. How the increased levels of this widely expressed nuclear structural component results a phenotype as specific as demyelination is a great mystery. This review summarizes what is currently known about the disease and describes recent work using animal and cell culture models that have provided critical insights into ADLD pathological mechanisms. The delineation of these pathways provides a fascinating glimpse into entirely novel roles for the nuclear lamina and will be critical for the identification of therapies for this fatal disease.
Keywords: chromatin; lamin B1; leukodystrophy; lipid synthesis; myelin; nuclear lamina; nuclear structure.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous